
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A digital workflow from crystallographic structure to single crystal particle attributes for predicting the formulation properties of terbutaline sulfate†
Thai T. H.
Nguyen
 *a,
Robert B.
Hammond
a,
Ioanna D.
Styliari
b,
Darragh
Murnane
b and
Kevin J.
Roberts
a
*a,
Robert B.
Hammond
a,
Ioanna D.
Styliari
b,
Darragh
Murnane
b and
Kevin J.
Roberts
a
aCentre for the Digital Design of Drug Products, School of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: H.Nguyen1@leeds.ac.uk
bSchool of Life and Medical Sciences, University of Hertfordshire, Hertfordshire AL10 9AB, UK
First published on 20th April 2020
Abstract
A detailed inter-molecular (synthonic) analysis of terbutaline sulfate, an ionic addition salt for inhalation drug formulation, is related to its crystal morphology, and through this to the surface chemistry of the habit faces and surface energy of the whole crystal. Coulombic interactions between the terbutaline cations and sulfate anions contribute 85% of the lattice energy, hydrogen bonding and dispersion interactions contribute 15%. Morphological prediction identifies a plate-like morphology composed of the forms {010}, {100}, {001} and {1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0} in good agreement with crystals grown from solution. Synthonic modelling of the intermolecular interactions, on a crystal face (hkl)-specific basis, reveals that the surface interactions on the forms with less, relative surface area: {100}, {001} and {10} manifest a greater surface energy, associated, notably, with a greater proportion of polar interactions at these surfaces compared to the morphological dominant {010} surfaces. The predicted total surface energies and their dispersive contributions are found to be in good agreement with those measured at high surface coverage using inverse gas chromatography (IGC). The modelling approach is complementary to IGC, as it enables surface sites of lower energy to be probed, that would require experimentally unachievable surface coverages. The utility of synthonic modelling, in understanding the surface properties of pharmaceutical materials, is highlighted through a workflow-based pathway for digital drug product design encompassing molecule structure, intermolecular packing, crystal morphology, surface energy and formulation properties.
0} in good agreement with crystals grown from solution. Synthonic modelling of the intermolecular interactions, on a crystal face (hkl)-specific basis, reveals that the surface interactions on the forms with less, relative surface area: {100}, {001} and {10} manifest a greater surface energy, associated, notably, with a greater proportion of polar interactions at these surfaces compared to the morphological dominant {010} surfaces. The predicted total surface energies and their dispersive contributions are found to be in good agreement with those measured at high surface coverage using inverse gas chromatography (IGC). The modelling approach is complementary to IGC, as it enables surface sites of lower energy to be probed, that would require experimentally unachievable surface coverages. The utility of synthonic modelling, in understanding the surface properties of pharmaceutical materials, is highlighted through a workflow-based pathway for digital drug product design encompassing molecule structure, intermolecular packing, crystal morphology, surface energy and formulation properties.
1. Introduction
Tert-Butyl [2-(3,5-dihydroxyphenyl)-2-hydroxyethyl] ammonium hemisulfate or terbutaline sulfate (TBS) is a β-adrenergic agonist: protonated terbutaline hemisulfate. TBS is present as the active pharmaceutical ingredient (API) in a number of commercial formulations for asthma therapy.1 The compound has five crystallographic forms2 including two anhydrous modifications (A and B), one monohydrate, one higher hydrate and one ethanoic acid solvate.3,4 The stable modification of TBS is an anhydrate B (Fig. 1) and it is this structure which is the subject of the current investigations. Two single-crystal structure determinations are reported for anhydrate B (ZIVKAQ5 and ZIVKAQ01 (ref. 6)) at 293 K which have similar R-factors (6.23% and 7.30%, respectively). Commercially, TBS is marketed as a racemic compound which decomposes on melting with the melting point being found to lie in the range of 258–260 °C.7 | ||
| Fig. 1 Molecular structure of the asymmetric unit of terbutaline sulfate anhydrous form B (ZIVKAQ).5 | ||
The commercial drug formulation typically contains respirable TBS drug particles that have a particle size of ∼1–5 μm, formulated with an α-lactose monohydrate carrier crystals having a particle size of 70–140 μm.8–10 Due to their small particle size, respirable API particles display highly cohesive and adhesive behaviour readily tending either to form agglomerates11 or to adhere to the surfaces of excipient materials, manufacturing process equipment or inhaler device components.12–15 Agglomerated or adhered drug particles must be re-dispersed effectively upon inhalation in order to be suitable for lung deposition. Hence, a detailed understanding of their surface chemistry and surface energy is important, in terms of determining the magnitude of inter-particulate interactions, for designing better formulations e.g. improving content uniformity, aerosolization and inhaled product performance.
Indeed, there have been relatively few studies of the effect of variations in the particle production processes upon surface crystallinity, particle adhesion and cohesion in terms of the inhalation performance of TBS.10,16,17 The work carried out so far has not provided any fundamental insight as regards the surface properties of TBS from a molecular-scale standpoint, particularly in terms of characterizing the external morphology, surface chemistry and particle surface energy. TBS is an interesting compound of generic relevance in the field of inhalation pharmaceutics. Detailed knowledge, provided by a fundamental intermolecular (synthonic) assessment, can deliver useful baseline data for improved understanding of inhalation-particle surface properties and associated inter-particle interactions.18
Accurate characterization of particle surface-energies can be challenging.19–21 For example, sessile-drop contact angle measurements on the individual facets of macroscopic single-crystals and/or powder compacts via the capillary rise method have not always been found to provide reliable results.22,23 Surface energy measurements from inverse gas chromatography (IGC) using both polar and apolar probes17,24–27 have, however, been found to be particularly useful in studies of the crystallinity, surface energy and surface properties of particles. Elsewhere, work using IGC has revealed a number of problematic issues such as sample preparation-dependency, large sample sizes (e.g. for micronised materials ∼50–100 mg and for coarse particles more than 2 grams of sample are required typically),28 time-utilisation and hence high cost. Variability in IGC measurements has been reported previously29–31 and the technique, even with finite dilution analysis, has not always been found to reliably assess the totality of a powder's surface area. The later reflects the technique's tendency to preferentially characterize the higher energy binding sites.32,33 Hence, if results are to be useful, there is a clear need for a better molecular-scale understanding of both the sources of variability and the impact of surface properties on IGC measurements.
Previous work by Grimsey et al.34 has shown the utility of molecular modelling in terms of understanding the surface chemistry of drug particles and also, its capability for identifying the interaction sites of molecular probes involved in IGC analysis. In this previous work, the micronized material was examined by making the assumption of particle breakage parallel to planes having the lowest surface attachment energies, i.e. the morphologically dominant crystal surfaces.35,36 This is despite the possibility that fracture could take place on non-crystal habit surfaces which might in turn have high surface energies and rugosity.37 Molecular dynamics simulations have also been employed to calculate the surface adsorption energies of the probe molecules,38 giving a reasonably close match to those determined from IGC. However, these studies were only carried out for the largest crystal surfaces, i.e. those which generally have the lowest surface energies. So far, no studies have adopted the holistic approach of integrating contributions from all the habit faces that comprise the external morphology of the crystal.
Synthonic modeling has been exploited extensively to investigate lattice energy, attachment energy and for the prediction of crystal morphologies using empirical force-field methods.39–41 The interior and surface properties of crystals can be predicted42,43 through calculations of the strength and direction of the inter-molecular interactions associated with the bulk (intrinsic synthons) and surface terminated (extrinsic synthons) structures.41,44–47 These approaches have been used to investigate a pharmaceutical salt46 and also to examine the cohesivity of APIs and excipients.18 However, acid addition salts (of which TBS is an example) have not, as of yet, been characterised despite their importance as APIs for inhalation drug formulation. Synthonic modelling tools can also predict the surface energy of a crystal and hence provide a clear link to surface energies measured by IGC.48,49 This modelling approach is also useful in being able to partition the calculated surface energy between the different contributing crystal surface planes (hkl) present on a fully facetted drug particle. Such attributes have obvious importance in terms of aiding the understanding of blending, flow and granulation for both APIs and excipients.
In summary, this paper reports an analysis of the key solid-state synthons, together with the predicted morphology, surface chemistry and surface energy of the morphologically important crystal surfaces of TBS. This analysis is complemented with calculations of solvent probe interactions with the TBS crystal habit surfaces to relate these both to the measured surface energies from IGC and to the surface chemistry that characterizes the crystal morphology.
2. Materials and methods
2.1 Materials
The molecular formula of TBS within the asymmetric unit is 2[C12H20NO3]+ SO42− with molecular mass, Mw = 548.65 g mol−1.5 The anhydrate form B of TBS crystallises in a triclinic structure with space group P with unit cell parameters a = 9.968 Å, b = 11.207 Å, c = 13.394 Å, α = 100.86°, β = 104.42°, γ = 101.63° with a tri-ionic (2 cation and 1 anion) asymmetric unit5 (see Fig. 1). In this work, the atomic coordinates were taken from the crystal structure5 with the lower R factor (refcode: ZIVKAQ).
For the experimental work; terbutaline sulfate was purchased from Sigma Aldrich (purity ≥98%) as a white to grey-white crystalline powder for crystallisation experiments. For surface energy measurements, terbutaline sulfate (batch AAEX) was kindly provided by AstraZeneca (Mölndal, Sweden), and was used as supplied.
2.2 Crystal growth
As received TBS was purified by recrystallization. TBS single crystals were prepared by cooling crystallisation as follows: 0.32 g of TBS were added to a mixture of 70% water and 30% ethanol (% by mass) to make up 1 g of solution. This was then heated to 70 °C and stirred for 30 minutes to ensure complete solute dissolution and then cooled to 5 °C and left for 2–3 days until crystallization occurred. An Olympus BX51 microscope, with a UMPlanFl 10×/0.30 objective was used to characterize the size and shape of the resultant crystals.2.3 Surface energy measurements
Specific surface area (SSABET) and surface energy (SE) analysis was conducted using a surface energy analyser (iGC-SEA, Surface Measurement Systems Ltd, UK; https://www.surfacemeasurementsystems.com/products/igc-sea/).0.247 g of terbutaline sulfate was packed into a silanised glass iGC column (internal diameter 4 mm). Prior to any measurements, the loaded column was conditioned using helium carrier gas at 10 scc min−1 for 2 h at 30 °C and 0% RH. Methane gas was injected at the start and the end of the experiments for the dead volume calculation. SSABET was calculated via Brunauer–Emmett–Teller (BET) theory, based on the n-octane adsorption isotherm data and using the peak max parameter.18
For the surface energy, the columns were equilibrated as mentioned above. Non-polar probes (n-decane, n-nonane, n-octane and n-heptane) and polar probes (chloroform, toluene, ethyl acetate and acetone) were injected into the column at concentrations consistent with a range of surface coverages (n/nm, between 0.5 up to 13% for all probes with the exception of n-decane that reached maximum surface coverage at 4.5%). Based upon an analysis method previously reported,50,51 the dispersive (γd, non-polar) and acid–base (γab, polar) components of the surface energy were calculated using the Dorris–Gray method and the peak centre of mass parameter. This is based on a linear fit on the 4 alkane probes for surface coverages 0.5–4.5% and based on 3 alkanes for surface coverages above 4.5%. Specific free energies of adsorption (ΔGSP) of the monopolar probes were obtained using the polarisation approach52 and the peak centre of mass parameter. All measurements were made in triplicate.
2.4 Computational modelling methodology
The computational analysis was carried out utilizing the Cambridge Crystallographic Data Centre (CCDC) materials Mercury,53 BIOVIA Materials Studio,54 HABIT98 (ref. 39) and SystSearch55,56 software. Partial atomic-charges were calculated using the semi-empirical quantum mechanics program MOPAC57 utilizing the Austin model 1(AM1) approach. The energetic strength and chemical identity of the intermolecular synthons, the lattice energy (Elatt or Ecr), and, hence, the slice (Esl) and attachment (Eatt) energies were calculated with HABIT98.39 This was aided by a detail analysis of the intermolecular interactions (synthons), both within the bulk structure and on the crystal habit faces, which was complemented by molecular-scale visualization of the bulk and surface chemistry. The digital workflow summarized in Fig. 2 provides an overview of the main features of the analysis used. | ||
| Fig. 2 A digital workflow from crystallographic structure to particle properties for the prediction of the formulation properties. | ||
 | (1) |
The lattice energy was calculated using spherical radii, for cut-off of the energy summation, between 5 and 50 Å, in 5 Å steps, to test and ensure convergence of the lattice energy. The calculation was repeated for each molecule in the asymmetric unit and the results were averaged over all sites in the asymmetric unit and then averaged over both the asymmetric units in the unit cell.
| Ecr = Ehklatt + Ehklsl | (2) |
| Rhkl ∼ Ehklatt | (3) |
The anisotropy factors for the crystal habit surfaces were calculated using eqn (4),
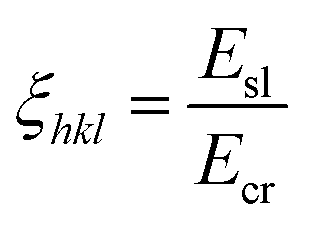 | (4) |
 | (5) |
The overall particle surface energy (γparticle)49 was calculated using eqn (6), as a surface area weighted average based on the attachment energy.
 | (6) |
3. Results and discussion
Results from the molecular and synthonic modelling analysis are described in sections 3.1 and 3.2, the morphological analysis and surface chemistry are presented in section 3.3 and 3.4, the comparison of the calculated surface energies with those measured using IGC is given in section 3.5 and the cross-correlation of the IGC data with intermolecular binding energies is given in section 3.6.3.1 Analysis of intermolecular packing within the bulk crystal structure
The intermolecular packing and crystal chemistry of TBS is summarised in Fig. 3 and Table 1.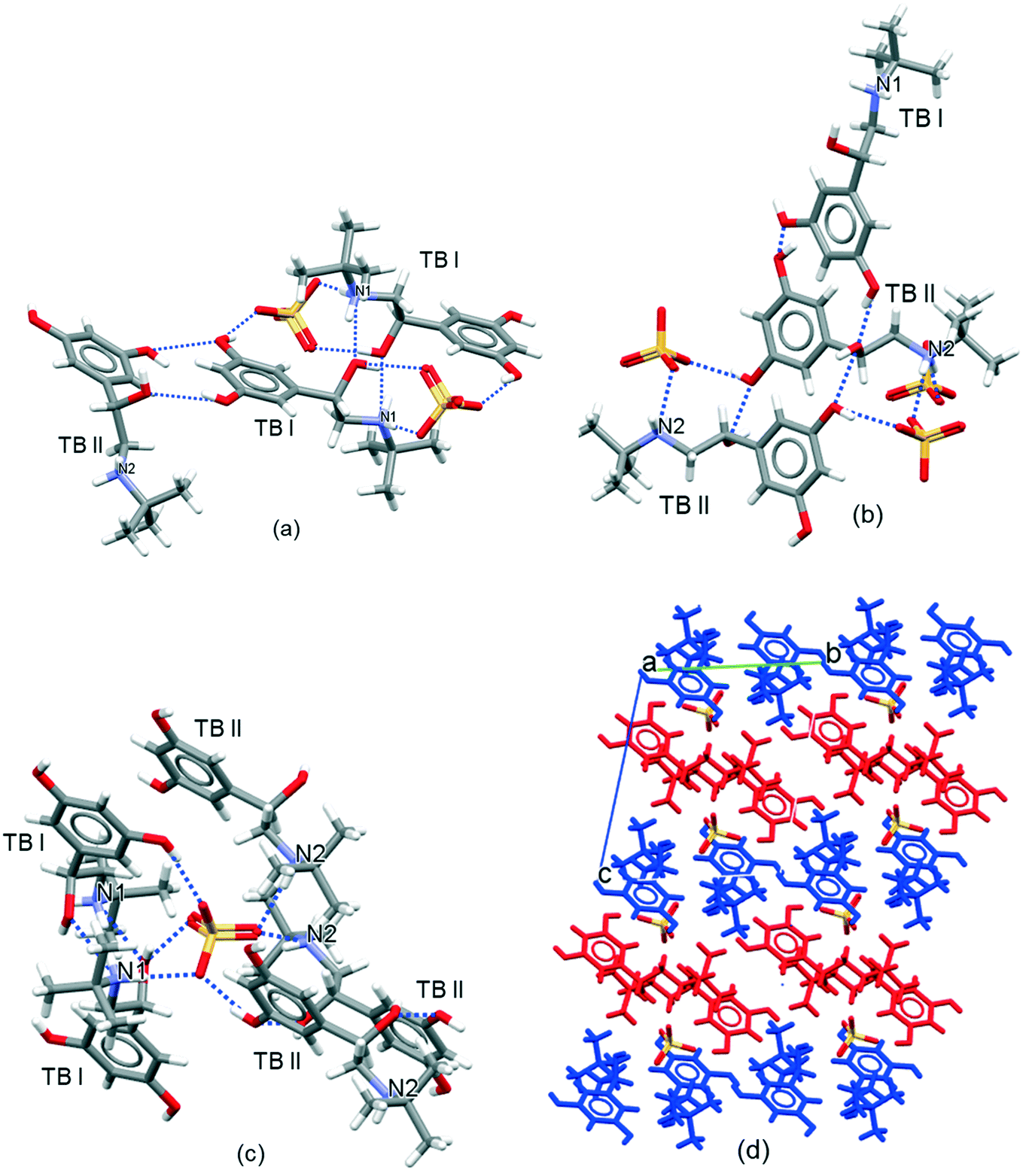 | ||
| Fig. 3 (a) Intermolecular coordination hydrogen-bonding shell for the TB I cation highlighting 7 H-bonding interactions with 4 other ions associated with cation TB I and cation TB II and 2 divalent sulfate counter anions; (b) intermolecular coordination hydrogen bonding shell for the TB II cation highlighting 7 H-bonding interactions with 5 other ions associated with cation TB I and cation TB II and 3 divalent sulfate counter anions; (c) intermolecular coordination hydrogen bonding shell for the SO42− group highlighting six H-bonds formed with 2 TB I and 3 TB II; (d) intermolecular packing showing terbutaline pack in alternate layers of terbutaline cation TB I (red) and cation TB II (blue). | ||
| Property | Molecular descriptor | Crystal descriptor | |||
|---|---|---|---|---|---|
| Two TB cations | Sulfate cation | TB I | TB II | Sulfate anion | |
| Number of hydrogen bond acceptors | 8 | 8 | 2 | 2 | 6 |
| Number of hydrogen bond donors | 10 | 0 | 5 | 5 | 0 |
The terbutaline cations have an asymmetric centre at carbon atom C7 and C19 for terbutaline cation 1 (TB I) and terbutaline cation 2 (TB II), respectively. The terbutaline cations consist of phenethylamine (C6H5–CH2–CH2–NH2–) groups in which the nitrogen atoms N1 and N2 are protonated (see Fig. 1). Each terbutaline consists of three alcohol groups (two –OH groups attached the benzene ring and one –OH group attached to the alkyl chain) and one amine –NH2 group. The asymmetric unit contains two terbutaline cations and one sulfate anion. The two crystallographically inequivalent terbutaline ions (TB I and TB II) exhibit differences in their conformation reflecting different rotations of the tertiary-butyl group about the connecting nitrogen to carbon covalent bond (NH2–C–(CH3)3 groups).
The basic unit of the crystal structure is an inversion-related terbutaline ion TB I dimer formed between the amine and alcohol groups attached to the alkyl chain. Each terbutaline cations TB I is bonded to one cation TB I, one cation TB II and two sulfate anions (Fig. 3a). Each cation TB II is bonded with one cation TB I and one cation TB II and 3 divalent sulfate anions (Fig. 3b).
In the crystal structure, each sulfate anion forms six hydrogen bonds with five TB cations (Fig. 3c) (two TB I and three TB II, one of the two, TB I, forms 2 hydrogen bonds with the sulfate group through N–H⋯O–S and O–H⋯O–S) utilizing six of the eight potential hydrogen bond acceptors from sulfate group. Each TB cation donates five (from three –OH groups and one –NH2 group in each TB cation) and accepts two hydrogen bonds (from two –OH groups).
In summary, there is a strong hydrogen bond network within the TBS crystal structure, with the use of all ten the hydrogen bond donors. However, not all the potential hydrogen bonding acceptor sites are used in the crystal structure, with four acceptors from TB I and TB II (two nitrogen atoms in the –NH2 group and two oxygens in the –OH group) and two acceptors from sulfate group are being unsaturated.
3.2 Lattice energy and identification of key bulk intrinsic synthons
Partial atomic-charges, calculated using the semi-empirical quantum mechanics program MOPAC, are provided in the section S1 in the (ESI†). Summation of the intermolecular interactions with distance revealed convergence at 15 Å to a lattice energy of −138.88 kcal mol−1 of asymmetric units with the coulombic interactions accounting for ∼85% of the lattice energy (Fig. 4). To the authors' knowledge, no sublimation enthalpy data has been published for TBS with which to compare to the calculated lattice energy. The summation of the percentage contribution to the lattice energy from the different molecular groups of the asymmetric unit, revealed that, TB I and TB II groups contributed 18.4% and 22.8% respectively, whilst the SO42− group was found to make the greatest contribution ca. 58.8% reflecting its intimate involvement with the strong coulombic anion–cation interactions.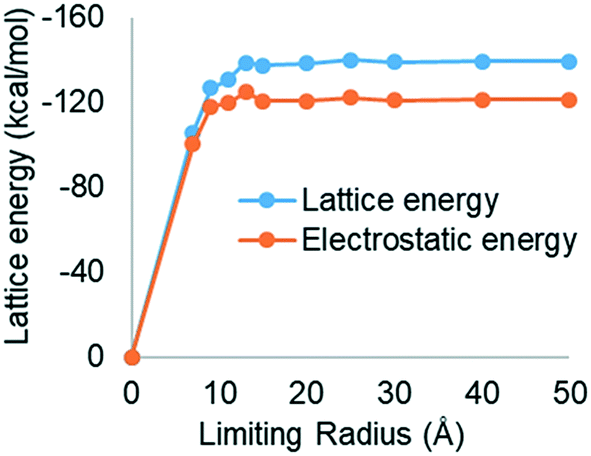 | ||
| Fig. 4 Lattice energy convergence as a function of limiting radius for TBS indicating that interactions at a distance greater than approximately 20 Å make a relatively small contribution to the lattice energy. | ||
Table 2 lists the top nine strongest, attractive intrinsic synthons found from the analysis of the bulk crystal structure and highlights the component of ions in the attractive synthons (with subscript a). All of the most attractive synthons were found to be between the sulphate anion – terbutaline cation pairs, indicating that the strongest interactions within crystal lattice are a result of the coulombic forces. Also, due to the symmetrical inequivalence of the terbutaline cation pairs, the synthon types found are quite similar to each other and simply occur between the different symmetrically inequivalent ions.
| Synthon | Ions involved | Inter-ion distance (Å) | Van der Waals contribution (kcal mol−1) | Coulombic contribution (kcal mol−1) | Total interaction (kcal mol−1) | {010} | {100} | {001} | {1−10} |
|---|---|---|---|---|---|---|---|---|---|
| A a | SF/TB II | 5.48 | −1.30 | −86.33 | −87.63 | SL | SL | SL | SL |
| B a | SF/TB II | 4.36 | −1.54 | −80.70 | −82.24 | SL | SL | SL | SL |
| C a | SF/TB I | 5.04 | −1.68 | −77.31 | −78.99 | SL | SL | ATT | SL |
| D a | SF/TB I | 4.81 | −0.89 | −74.39 | −75.28 | SL | SL | SL | SL |
| E a | SF/TB I | 6.56 | −0.31 | −48.14 | −48.45 | SL | ATT | SL | ATT |
| F a | SF/TB II | 8.57 | −0.17 | −41.20 | −41.37 | ATT | SL | SL | ATT |
| G a | SF/TB II | 7.50 | −0.77 | −38.23 | −39.00 | SL | ATT | SL | ATT |
| H a | SF/TB II | 9.66 | −0.03 | −36.77 | −36.80 | SL | SL | SL | SL |
| I a | SF/TB II | 12.31 | −0.01 | −32.50 | −32.51 | SL | SL | ATT | SL |
| A r | SF/SF | 6.43 | −0.10 | 106.96 | 106.86 | SL | SL | SL | SL |
| B r | SF/SF | 8.19 | −0.02 | 81.20 | 81.18 | SL | SL | ATT | SL |
| C r | SF/SF | 9.26 | −0.01 | 71.93 | 71.92 | SL | ATT | ATT | ATT |
| D r | SF/SF | 9.47 | 0.00 | 70.05 | 70.05 | ATT | SL | SL | ATT |
Fig. 5 reveals that the orientation of specific atomic groups, particularly the –OH and –NH2 functional groups strongly influences the strength of the synthon. Synthons Aa, Ba, Ca and Da (interactions between sulfate with TB I or TB II) have the closest orientation of the oppositely charged SO42− and –NH2+, resulting in a very strong coulombic interaction. For synthon Aa and Ba (interaction between sulfate and TB II), the H-bonding is formed due to the close proximity of the donor H on the NH2+ group and the acceptor O on the SO42− group. For the synthon Ca and Da (interaction between sulfate and TB I) the H-bonding is formed due to the close proximity of the donor hydrogen atoms on the NH2+ and –OH group and acceptor oxygen atoms on the SO42− group. The added attractive force of these H-bonds probably contributes to this synthon being calculated to be the most attractive interaction within the structure. It was also observed, as expected that, in general, as the distance between the SO42− and NH2+ groups increases, the attractive force between the ions decreases; for example distance between sulfur of SO42− and nitrogen NH2+ of synthon Aa and Ba decrease from 5.48 to 4.36 Å; the attractive force increases −2.05 to −2.59 kcal mol−1. However, although the SO42− and the NH2+ groups are closer in synthon La, synthons Ia/Ka and Ja were found to have a greater attractive interaction due to these synthons forming more significant van der Waals and H-bonding interactions, respectively.
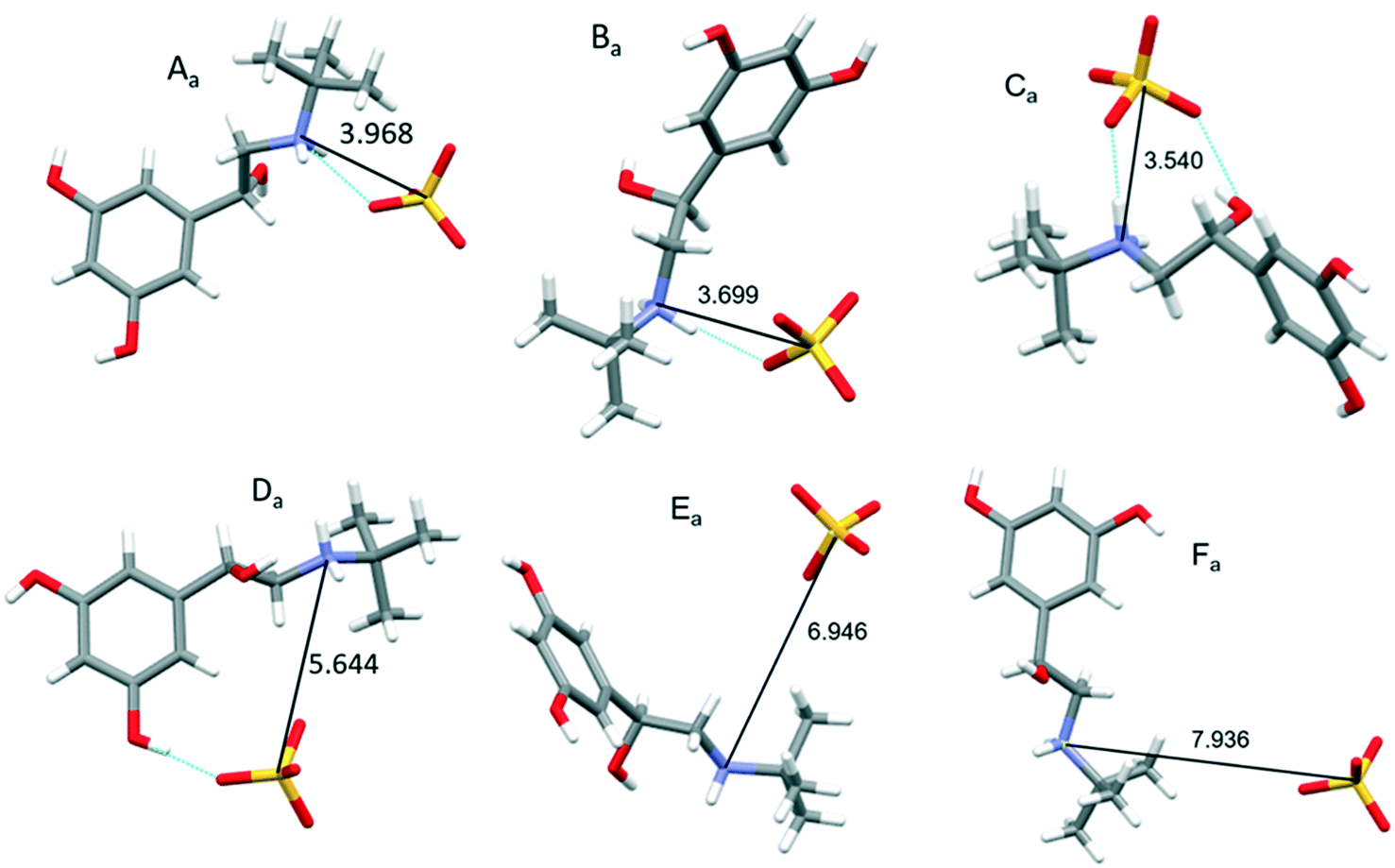 | ||
| Fig. 5 Molecular orientation of the top six (Aa, Ba, Ca, Da, Ea and Fa) attractive synthons shown in Table 2 indicating that the major interactions are between the oppositely charged ions. Similar interaction types that are only different because they are between the different terbutaline cations type I and type II all distances are quoted between S in SO42− and N in NH2+. The dotted blue lines indicate the hydrogen bonds. | ||
The strongest repulsions within the crystal structure were found to be between the sulfate anions (see ESI,† section S2). Due to the symmetrical equivalence of the anions, the four most-repulsive, independent pairs only are illustrated. Table 2 lists that, as expected, the majority of the repulsion energy is due to the coulombic repulsions. These strongest repulsive synthons were found to be between 6.43–9.48 Å apart (S–S). These repulsions were more than compensated for by the strong attractive forces between the anion–cation pairs.
3.3 Predicted morphology and identification of key surface-terminated extrinsic synthons
The predicted morphology, shown in Fig. 6(a) and detailed in Table 3 column 1–6 based on the attachment energy model, shows a plate-like morphology with the {010} being the dominant form, along with the smaller {100}, {001} and {10} surfaces. The predicted morphology agrees well with the observed experimental crystal morphology of TBS crystals grown in a mixture of 70% water and 30% ethanol at 5 °C (see Fig. 6(b)).
 | ||
| Fig. 6 Predicted morphology using attachment energy model showing a plate-like crystal morphology with the {010} being the dominant form, along with the smaller {100}, {001} and {10} surfaces (a) and experimentally observed morphology of TBS single crystals (b). | ||
| Crystal face | Multiplicity | Interplannar spacing dhkl (Angstrom) | Surface area Ahkl (%) | Slice energy Esl (kcal mol−1) | Attachment energy Eatt (kcal mol−1) | Surface saturation ζhkl (%) | Dispersive surface energy (mJ m−2) | Total surface energy (mJ m−2) |
|---|---|---|---|---|---|---|---|---|
| {010} | 2 | 10.62 | 26.33 | −133.72 | −5.16 | 96.28 | 23.24 | 27.73 |
| {100} | 2 | 9.31 | 10.29 | −128.15 | −10.73 | 92.27 | 31.44 | 50.57 |
| {001} | 2 | 12.55 | 8.69 | −121.71 | −17.16 | 87.64 | 38.99 | 108.97 |
| {10} |
2 | 8.14 | 4.70 | −127.81 | −11.07 | 92.03 | 29.61 | 45.57 |
The percentage surface saturation (anisotropy factors) are listed in Table 3 column 7. As expected, the {010} surface contains the highest degree of saturation, 96.28%, due to fewer strong synthons being broken at the surface. In comparison, the {100}, {001} and {10} surfaces have 92.3%, 87.6% and 92.03% saturation, respectively; as they are predicted to have strong anion/cation synthons broken at their respective surfaces. This agrees with the dominant faces rank order {10} < {001} < {100} < {010}.
In this study the synthons that are predicted to contribute to the attachment energy, and hence growth, of a given crystal surface have been identified and displayed in Table 2. This reveals that stronger intermolecular interactions (synthons) contributing to the attachment energy Eatt of the crystal faces {hkl} leads to the higher growth rates of {hkl}. The strongest interaction contributing to the attachment energy at the {010} surface was found to be synthon Fa (−41.37 kcal mol−1) (see Table 2), which is the coulombic interaction between sulfate and amine group of terbutaline cation 2. The strongest interactions contributing to the attachment energy at the {10} surface were found to be Ea, Fa, and Ga (−48.45, −41.37 and −39.00 kcal mol−1). These predictions are consistent with the morphological dominance of this face and with the order of the importance of the faces: {10} < {001} < {100} < {010}.
3.4 Analysis of the surface chemistry of the habit planes
The morphology of a crystal in different growth environments depends, collectively, on the relative growth rates associated with its constituent crystal-habit faces. Ultimately these rates are linked to the respective interaction energies associated with the strongest synthons on these surfaces. The growth morphology in a vacuum of an individual habit face can be rationalised in terms of the respective molecular arrangements, and hence synthon orientation, on these specific faces. Hence, molecular modelling can be utilised to examine the surface chemistry of the {010}, {100}, {001} and {10} surfaces. For the faces listed in Table 3, the extrinsic synthons identified as contributing to the attachment energies were labelled on molecular models of the surface as shown in the section S3 in the ESI.†
The {010} form exhibits one –OH group from each terbutaline cation exposed on the surfaces with all the H-bonding being embedded within crystal surfaces. The surfaces of the forms {100} and {001} exhibit two –OH groups one each from each terbutaline cation exposed on the surfaces; however, the {001} surfaces have non-bonded H-bond acceptor groups across the surface which are available to form hydrogen bonds with probe molecules. The {10} morphological form exposes three –OH groups and the protonated –NH2–C3H9 moiety on the crystal surface. This indicates that these faces are more polar than the other faces and explains the ranking of the morphological importance of the exposed crystal surfaces in terms of surface area.
3.5 Comparison of predicted cumulative particle surface energies with measured IGC data
The surface energies, as a function of surface plane, together with the fractional surface areas of the associated habit plane are given in Table 3, columns 9 and 4, respectively. The predicted cumulative surface energies and the measured surface energies of non-micronized TBS, as measured by IGC, are reported in Table 4 and Fig. 7. The dispersive component of the surface energy measured by IGC ranges between 49.1 and 32.8 mJ m−2; this is comparable to measured values of micronized and supercritical processed TBS reported by Rehman, M. et al.71 (58.61 ± 0.3 for the micronized and 55.05–57.37 for the supercritical processed material).| Surface energy | Dispersive SE (mJ m−2) | Total SE (mJ m−2) |
|---|---|---|
| Calculated surface energy (weighting with % surface area) (mJ m−2) | 28.27 | 48.2 |
| Surface energy measured by IGC for surface coverage [0.5–13%] | 49.1–32.8 | 103.3–64.7 |
 | ||
| Fig. 7 Relative pressure reached during the IGC experiments as a function of the surface coverage for the alkane probes (a); measured dispersive and total surface energy of non-micronized terbutaline sulfate supplied by AZ using IGC showing the surface energy decrease with increasing surface coverage (b). | ||
The predicted surface energy showed significant differences between the surface energies of individual crystal surfaces (see Table 3, columns 8 and 9). The surface energies of the {10} and {001} forms are greatest whilst the surface energy of the {010} form is smallest. This agrees with the surface chemistry analysis which indicates that the {10} and {001} forms have more polar and non-polar components exposed on the surfaces contributing to the dispersive and polar surface energy, for example, for the {10} faces the –CH3–CH3–CH3 and –CH2 group (alkyl groups) attached to the –NH2 contribute to dispersive energy components and –NH3 and three –OH groups contribute to polar surface energy components.
The predicted surface energy, which is based upon an idealised perfect termination of the bulk crystal lattice, gave smaller values than those measured for TBS supplied from AZ (see Table 4) and those measured for micronized and processed TBS reported by Rehman, M. et al.71 It has been reported that IGC performed in the infinite dilution regime usually yields higher values of dispersive surface energy than other measurement techniques,72 as well as sometimes providing an unrepresentative measure of the majority of the powder surface area. Infinite dilution IGC, historically performed at 0.03 p/p0, has been shown to probe the crystal surface regions with the highest energy, while finite dilution IGC can provide a surface heterogeneity map.32,73–75 The effectiveness of infinite dilution IGC is likely due to adsorbed molecules having no other adsorbate molecules close enough to allow any adsorbate–adsorbate interactions, hence there is a lack of competition for adsorbate binding sites. The highest energetic sites probed by infinite dilution IGC may not dominate the inter-particulate contact areas, since their relative contribution to the total particle surface area may be exceedingly low. For example, for TBS the {001} faces contribute the highest surface energy (Table 3), but this face only provides 8.69% of the crystal surface area (Table 3).
It was previously reported33,76 that size reduction, such as micronization, leads to an increase in the measured dispersive surface energy of the drug substance with increasing extent of micronization. The increase in surface energy is probably due to the generation of new, higher energy sites of crystalline disorder due to impaction events or particle fracture.33 Infinite dilution IGC measurement of surface energy would therefore be expected to be dominated by the latter, unrepresentative high energy sites.
It is significant that computational prediction of surface energy for the highest energy sites (the {001} surfaces: 109 mJ m−2) correlated well with the experimental measurements at low surface coverage (103.3 mJ m−2), where the highest energy surface sites are probed (both from the standpoint of crystallography and process-induced disorder). It was evident that the surface energy values determined experimentally for the highest surface coverage (which should probe the relatively lower surface energy sites) showed an improved correlation with the average calculated surface energy when this is weighted with respect to the relative contribution of each crystal face to the total surface area of the crystal (see Table 4).
3.6 Comparison of the predicted adsorption energy for the different polar probes with those measured using IGC
The results for the grid-search modelling are given in Fig. 8. These provide histograms of calculated interaction energies for a series of probe vapours (chloroform, toluene, acetone, and ethyl acetate) on TBS crystal surfaces {010}, {100}, {001} and {10}. The strongest interaction energies, the mean interaction energy distribution of a probe molecule on TBS crystal surfaces, together with the total interaction energy of probe molecules on the TBS particles are summarized in Table 5.
 | ||
| Fig. 8 Histogram of interaction energy distribution of (a) ethyl acetate, (b) chloroform, (c) acetone and (d) toluene of crystal surfaces of {010} (black), {100} (red), {001} (green) and {10} (blue) of TBS showing binding energies of probe molecules are different on individual crystal surfaces (with bin size 2 kJ mol−1). | ||
| Maximum and mean interaction energies of a probe molecule on TBS crystal surfaces (kJ mol−1) | Total interaction energy weighted with % surface area of individual crystal surfaces (kJ mol−1) | Surface free energies of adsorption (−ΔGSP) (kJ mol−1) measured by IGC | |||||
|---|---|---|---|---|---|---|---|
| Probe molecules | {010} | {100} | {001} | {10} |
0.5% surface coverage | 13% surface coverage | |
| Ethyl acetate | |||||||
| Maximum | −27.68 | −33.49 | −46.73 | −52.16 | −34.49 | 15.85 ± 0.04 | 11.17 ± 0.45 |
| Mean | −10.71 | −10.06 | −15.28 | −13.78 | −12.17 | ||
| Chloroform | |||||||
| Maximum | −18.65 | −28.38 | −21.31 | −30.55 | −22.24 | 7.88 ± 0.03 | 5.61 ± 0.36 |
| Mean | −9.83 | −11.09 | −11.61 | −12.74 | −10.68 | ||
| Acetone | |||||||
| Maximum | −24.93 | −32.09 | −40.55 | −43.02 | −30.82 | 17.02 ± 0.05 | 11.19 ± 0.5 |
| Mean | −10.34 | −11.94 | −15.43 | −13.94 | −11.89 | ||
| Toluene | |||||||
| Maximum | −18.91 | −25.83 | −24.22 | −29.20 | −22.41 | 4.58 ± 0.02 | 3.77 ± 0.16 |
| Mean | −10.23 | −11.22 | −11.77 | −12.59 | −10.93 | ||
This study elucidates the anisotropic surface chemistry of the major faces of TBS, identifies that polar solvent-probe molecules were found to interact more strongly with the side faces {100}, {10} and {001} and least with the large {010} surface. This illustrates that the relative contribution of a crystallographic face to its total surface area plays an important role in the preferential binding site for the probe molecules. Indeed, the modelling studies indicated that the surface roughness at the molecular-level can have a major influence on the probe molecule adsorption energies, which may impact the actual process of adsorption in experimental determinations. This increased surface roughness can be expected to provide a favourable docking site for adsorption in a surface ‘valley’, for the probe molecules. Hence, it has been shown that the higher interaction energy sites for the {100}, {001} and {10} surfaces tend to be located in the surface ‘valleys’. Particularly for the {10} surface, the probe molecules prefer to bind in a channel present at the crystal surfaces, which is also the case for ethyl acetate, chloroform and toluene probe molecules, resulting in high interaction energies at this surface. Further details for the preferred binding position of the probe molecules on the TBS crystal surfaces are presented in the ESI† (see section S4).
The data analysis also elucidated that for toluene and chloroform, the solvent probe interactions with all the surfaces are found to be almost completely from dispersive force contributions and coulombic interactions (for chloroform), without a hydrogen bonding contribution. However, it should be noted that in a real solvent system, chloroform is known to form weak hydrogen bonds, the Dreiding forcefield used in these calculations does not consider chlorine as a hydrogen bond acceptor atom and hence no hydrogen bonding contribution would be expected. Similarly, toluene as a non-polar solvent molecule, is incapable of forming hydrogen bonds so would also be expected to have zero contribution to the overall interaction energy. In contrast, for ethyl acetate and acetone, the interaction energies are found to have had a significant H-bonding contribution and, additionally, the coulombic energy contribution is significant, particularly for acetone.
The measured specific surface energies ΔGSP (kJ mol−1) using chloroform, toluene, ethyl acetate, and acetone are given in Fig. 9.
 | ||
| Fig. 9 Relative pressure reached during the IGC experiments as a function of the surface coverage for the polar probes (a); the measured surface free energies of adsorption ΔGSP (kJ mol−1) using chloroform, toluene, ethyl acetate, and acetone (b). | ||
Despite the poorer agreement, a trend of decreasing measured surface energy was apparent as surface coverage was increased, probing the lower surface energy sites (see Fig. 7b and 9b). The trend for surface energy in Fig. 9 indicates that the surface energy distribution of TBS is quite heterogeneous. During the finite dilution IGC experiments, the surface energy distribution was calculated via the retention volumes of the probes at a specific surface coverage in order to allow for the differences between the various probes. However, this is also a limitation of the technique as the maximum injectable amount – and as such the maximum surface coverage a selected probe can achieve – is determined by the probe's cross-sectional area. In this present work, it was not possible to measure the dispersive component of TBS surface energy accurately above surface coverages of 13% as that would reduce the number of available alkane probes to two (octane and heptane), insufficient for accurate linear regression. Further analysis of the adsorption isotherms of the probes (see Fig. 7a and 9a, and section S5 in the ESI† for the adsorption isotherms) showed that even at the surface coverage of 13%, the partial pressure in the column did not exceed 0.06 P/P0 which indicates that a full monolayer coverage has not been reached and thus only a fraction of the heterogeneous surface was probed. Consequently, this would suggest that the experimental surface energy measurements by IGC did not achieve sufficient surface coverage to probe the lowest surface energy sites in the same way as have been predicted in the molecular modelling-based grid search approach.
4. Conclusions
In this work, molecular and synthonic modelling tools have been successfully applied to link dominant synthon contributions, the percentage surface saturation, the surface area, surface chemistry analysis (functional groups exposed on crystal surfaces, extrinsic synthons) surface energy and physical properties. Synthonic modeling revealed the extent of the importance of coulombic interactions between the salt pairs to stabilize the lattice energy, compared with small, free base, molecule crystal structures. Attachment energy morphology predictions produced a plate-like morphology with the {010} being the dominant form, along with the smaller {100}, {10}, {101} and {001} forms, which is consistent with the crystal morphology observed from solution grown crystals. The synthon analysis explains why the relative morphological importance to the crystal morphology of forms is in the order {10} < {001} < {100} < {010}. The faster growing {10} surfaces were predicted to have many more of the strong synthons notably Ea, Fa and Ga contributing to their attachment energy.
The calculated dispersive and total surface energy had lower values than the measured data reported in this study and tended to correlate better at a surface coverage of 13% compared to 0.5% surface coverage. This might be due to the experimental surface energy measurements by IGC in this study which did not achieve sufficient surface coverage to probe all the lowest surface-energy sites, combined with the finding that the surface energy distribution of TBS across its different crystal habit faces is quite heterogeneous. From the calculated adsorption energy for the different polar probes on TBS crystals, the mean interaction energy was found to provide a better correlation with the measured surface free energies of adsorption (−ΔGSP).
The calculated surface energy analysis was found to be very helpful for assessing and interpreting the measured IGC data, notably through its ability to both partition surface energies between the different morphological forms as well as probe lower energy binding sites not routinely accessible using IGC, for designing formulation processes for inhalation drugs. This is due to the importance of surface properties, their link to drug particulate-interactions and the associated, intrinsic, powder properties needed for achieving effective drug aerosolization, efficacy, lung delivery and therapeutic success. This suggests that the type of analysis presented here might have more general applications in the formulation design of other APIs in order to understand, in-detail, the fundamental material behavior at the molecular scale.
List of symbols
| E latt | Lattice energy |
| E sl | Slice energy |
| E att | Attachment energy |
| ξ hkl | Anisotropy factors |
| Z | The number of molecules per unit cell |
| d hkl | The thickness of the growth step layer |
| N A | Avogadro's constant |
| V | The unit cell volume |
| γ | Surface energy |
| γ d | Dispersive components of surface energy (non-polar) |
| γ ab | Polar components of the surface energy (acid–base) |
| ΔGSP | Specific free energies of desorption |
Glossary of terms
| vdW | Van der Waals |
| H-Bonding | Hydrogen bonding |
| Synthons | Pairwise intermolecular interactions |
| Intrinsic synthons | Fully saturated synthons found in the bulk of the crystal structure |
| Extrinsic synthons | Unsaturated synthons due to surface termination of the crystal structure |
| Lattice energy | Strength of the intermolecular interactions within the crystal structure per mol |
| Slice energy | Energy of intermolecular interactions found within one d-spacing on the (hkl) crystallographic plane |
| Attachment energy | Energy of intermolecular interactions formed when a crystal slab one d-spacing thick, defined by (hkl) plane, is inserted into a bulk crystal structure |
| Anisotropy factor | The degree of saturation of a molecule exposed at a cleaved crystal surface (hkl), in comparison to the same molecule fully saturated in the bulk structure |
Conflicts of interest
The authors declare no competing finance interest.Acknowledgements
This work was supported by the UK's EPSRC research grant EP/N025075/1 INFORM2020 “Molecules to manufacture: Processing and Formulation Engineering of Inhalable Nanoaggregates and Micro-particles”. This work also builds upon research on morphological modelling supported by EPSRC grant ‘HABIT – Crystal morphology from crystallographic and growth environment factors’ through EPSRC grant EP/I028293/1 and the Synthonic Engineering programme supported by Pfizer, Boeringer-Ingellheim, Novartis and Syngenta. We are also grateful to Dr. Kyrre Thalberg from Astra Zeneca (Sweden) for providing terbutaline sulfate for this study.References
- S. P. Newman, F. Morén, E. Trofast, N. Talaee and S. W. Clarke, Int. J. Pharm., 1991, 74, 209–213 CrossRef CAS.
- R. K. Harris, P. Hodgkinson, T. Larsson, A. Muruganantham, I. Ymén, D. S. Yufit and V. Zorin, Cryst. Growth Des., 2008, 8, 80–90 CrossRef CAS.
- L. Eriksson, P. L. Wang and P. Werner, Z. Kristallogr., 1995, 210, 891 CAS.
- D. C. Apperley, A. S. Batsanov, S. J. Clark, R. K. Harris, P. Hodgkinson and D. B. Jochym, J. Mol. Struct., 2012, 1015, 192–201 CrossRef CAS.
- R. Sengupta and J. Dattagupta, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 162–164 CrossRef.
- L. Eriksson, P. L. Wang and P. E. Werner, Z. Kristallogr. – New Cryst. Struct., 1997, 212, 267 CAS.
- S. Ahuja and J. Ashman, in Analytical Profiles of Drug Substances, ed. K. Florey, Academic Press, 1990, vol. 19, pp. 601–625 Search PubMed.
- G. Pilcer, N. Wauthoz and K. Amighi, Adv. Drug Delivery Rev., 2012, 64, 233–256 CrossRef CAS PubMed.
- X. M. Zeng, K. H. Pandhal and G. P. Martin, Int. J. Pharm., 2000, 197, 41–52 CrossRef CAS.
- B. Dickhoff, A. De Boer, D. Lambregts and H. Frijlink, Eur. J. Pharm. Biopharm., 2003, 56, 291–302 CrossRef CAS PubMed.
- S. Adi, H. Adi, H.-K. Chan, W. H. Finlay, Z. Tong, R. Yang and A. Yu, J. Aerosol Sci., 2011, 42, 285–294 CrossRef CAS.
- J. C. Hooton, M. D. Jones and R. Price, J. Pharm. Sci., 2006, 95, 1288–1297 CrossRef CAS.
- S. Karner and N. Anne Urbanetz, J. Aerosol Sci., 2011, 42, 428–445 CrossRef CAS.
- G. Rowley, Int. J. Pharm., 2001, 227, 47–55 CrossRef CAS PubMed.
- D. Morton, US Pat. Application No. 11/791, 2008, p. 385 Search PubMed.
- T. H. H. Thi, F. Danède, M. Descamps and M.-P. Flament, Eur. J. Pharm. Biopharm., 2008, 70, 380–388 CrossRef CAS PubMed.
- S. C. Das, I. G. Tucker and P. J. Stewart, Curr. Pharm. Des., 2015, 21, 3932–3944 CrossRef CAS PubMed.
- V. Ramachandran, D. Murnane, R. B. Hammond, J. Pickering, K. J. Roberts, M. Soufian, B. Forbes, S. Jaffari, G. P. Martin and E. Collins, Mol. Pharmaceutics, 2014, 12, 18–33 CrossRef PubMed.
- P. Begat, D. A. Morton, J. N. Staniforth and R. Price, Pharm. Res., 2004, 21, 1591–1597 CrossRef CAS PubMed.
- C. J. Roberts, Eur. J. Pharm. Sci., 2005, 24, 153–157 CrossRef CAS PubMed.
- D. R. Williams, Curr. Pharm. Des., 2015, 21, 2677–2694 CrossRef CAS PubMed.
- J. Y. Y. Heng, A. Bismarck and D. R. Williams, AAPS PharmSciTech, 2006, 7, E12–E20 CrossRef PubMed.
- D. Zhang, J. H. Flory, S. Panmai, U. Batra and M. J. Kaufman, Colloids Surf., A, 2002, 206, 547–554 CrossRef CAS.
- D. Cline and R. Dalby, Pharm. Res., 2002, 19, 1274–1277 CrossRef CAS PubMed.
- D. Traini, P. Rogueda, P. Young and R. Price, Pharm. Res., 2005, 22, 816–825 CrossRef CAS PubMed.
- M. D. Jones, P. Young and D. Traini, Adv. Drug Delivery Rev., 2012, 64, 285–293 CrossRef CAS PubMed.
- H. H. Tong, B. Y. Shekunov, P. York and A. H. Chow, J. Pharm. Sci., 2006, 95, 228–233 CrossRef CAS PubMed.
- E. Hadjittofis, G. G. Zhang and J. Y. Heng, RSC Adv., 2017, 7, 12194–12200 RSC.
- V. Swaminathan, J. Cobb and I. Saracovan, Int. J. Pharm., 2006, 312, 158–165 CrossRef CAS PubMed.
- G. Garnier and W. G. Glasser, J. Adhes., 1994, 46, 165–180 CrossRef CAS.
- S. P. Chamarthy, R. Pinal and M. T. Carvajal, AAPS PharmSciTech, 2009, 10, 780 CrossRef CAS PubMed.
- R. Ho and J. Y. Heng, KONA Powder Part. J., 2013, 30, 164–180 CrossRef CAS.
- R. Ho, M. Naderi, J. Y. Heng, D. R. Williams, F. Thielmann, P. Bouza, A. R. Keith, G. Thiele and D. J. Burnett, Pharm. Res., 2012, 29, 2806–2816 CrossRef CAS PubMed.
- I. M. Grimsey, J. C. Osborn, S. W. Doughty, P. York and R. C. Rowe, J. Chromatogr. A, 2002, 969, 49–57 CrossRef CAS PubMed.
- C. C. Sun and Y. H. Kiang, J. Pharm. Sci., 2008, 97, 3456–3461 CrossRef CAS PubMed.
- M. Bryant, A. Maloney and R. Sykes, CrystEngComm, 2018, 20, 2698–2704 RSC.
- D. Olusanmi, K. J. Roberts, M. Ghadiri and Y. Ding, Int. J. Pharm., 2011, 411, 49–63 CrossRef CAS PubMed.
- A. Saxena, J. Kendrick, I. Grimsey and L. Mackin, Int. J. Pharm., 2007, 343, 173–180 CrossRef CAS PubMed.
- G. Clydesdale, K. Roberts and R. Docherty, J. Cryst. Growth, 1996, 166, 78–83 CrossRef CAS.
- G. Clydesdale, K. J. Roberts, G. B. Telfer and D. J. Grant, J. Pharm. Sci., 1997, 86, 135–141 CrossRef CAS PubMed.
- T. T. Nguyen, I. Rosbottom, I. Marziano, R. B. Hammond and K. J. Roberts, Cryst. Growth Des., 2017, 17, 3088–3099 CrossRef CAS.
- J. Pickering, R. B. Hammond, V. Ramachandran, M. Soufian and K. J. Roberts, in Engineering Crystallography: From Molecule to Crystal to Functional Form, ed. K. J. Roberts, R. Docherty and R. Tamura, Springer Advanced Study Institute (ASI) Series, 2017, pp. 155–176, ISBN 878-194-024-1118-1118/1115-1117/1117-1111 Search PubMed.
- K. J. Roberts, R. B. Hammond, V. Ramachandran and R. Docherty, in Computational Pharmaceutical Solid State Chemistry, ed. Y. A. Abramov, Copyright © 2015 Wiley, Inc., 2016, pp. 175–210 Search PubMed.
- R. Docherty and K. Roberts, J. Cryst. Growth, 1988, 88, 159–168 CrossRef CAS.
- T. D. Turner, L. E. Hatcher, C. C. Wilson and K. J. Roberts, J. Pharm. Sci., 2019, 108, 1779–1787 CrossRef CAS PubMed.
- A. A. Moldovan, I. Rosbottom, V. Ramachandran, C. M. Pask, O. Olomukhoro and K. J. Roberts, J. Pharm. Sci., 2017, 106, 882–891 CrossRef CAS PubMed.
- I. Rosbottom, K. Roberts and R. Docherty, CrystEngComm, 2015, 17, 5768–5788 RSC.
- R. B. Hammond, K. Pencheva and K. J. Roberts, Cryst. Growth Des., 2006, 6, 1324–1334 CrossRef CAS.
- S. N. A. Yusop, PhD Thesis, University of Leeds, 2014 Search PubMed.
- S. Jaffari, B. Forbes, E. Collins, J. Khoo, G. P. Martin and D. Murnane, Pharm. Res., 2014, 31, 3251–3264 CrossRef CAS PubMed.
- S. Jaffari, B. Forbes, E. Collins, D. J. Barlow, G. P. Martin and D. Murnane, Int. J. Pharm., 2013, 447, 124–131 CrossRef CAS PubMed.
- S. Dong, M. Brendle and J. Donnet, Chromatographia, 1989, 28, 469–472 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury 4.2.1, The Cambridge Crystallographic Data Centre (CCDC), Cambridge, 2019 Search PubMed.
- Dassault Systèmes BIOVIA, Material Studio (17.1.0.48), Dassault Systèmes, San Diego, 2017 Search PubMed.
- R. Hammond, K. Pencheva and K. Roberts, Cryst. Growth Des., 2007, 7, 875–884 CrossRef CAS.
- R. Hammond, S. Jeck, C. Ma, K. Pencheva, K. Roberts and T. Auffret, J. Pharm. Sci., 2009, 98, 4589–4602 CrossRef CAS PubMed.
- J. J. Stewart, J. Comput.-Aided Mol. Des., 1990, 4, 1–103 CrossRef CAS PubMed.
- M. Clark, R. D. Cramer III and N. Van Opdenbosch, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- A. Gellé and M.-B. Lepetit, J. Chem. Phys., 2008, 128, 244716 CrossRef PubMed.
- G. Wulff, Z. Kristallogr. Cryst. Mater, 1901, 34, 449–530 CAS.
- P. Hartman and W. Perdok, Acta Crystallogr., 1955, 8, 49–52 CrossRef.
- P. Hartman and P. Bennema, J. Cryst. Growth, 1980, 49, 145–156 CrossRef CAS.
- L. Jetten, H. Human, P. Bennema and J. Van Der Eerden, J. Cryst. Growth, 1984, 68, 503–516 CrossRef CAS.
- H. Human, J. Van Der Eerden, L. Jetten and J. Odekerken, J. Cryst. Growth, 1981, 51, 589–600 CrossRef CAS.
- P. Bennema and J. Van der Eerden, J. Cryst. Growth, 1977, 42, 201–213 CrossRef CAS.
- R. B. Hammond, K. J. Roberts, E. D. L. Smith and R. Docherty, J. Phys. Chem. B, 1999, 103, 7762–7770 CrossRef CAS.
- R. B. Hammond, K. Pencheva and K. J. Roberts, Cryst. Growth Des., 2007, 7, 875–884 CrossRef CAS.
- R. B. Hammond, C. Ma, K. J. Roberts, P. Y. Ghi and R. K. Harris, J. Phys. Chem. B, 2003, 107, 11820–11826 CrossRef CAS.
- R. B. Hammond, R. S. Hashim, C. Ma and K. J. Roberts, J. Pharm. Sci., 2006, 95, 2361–2372 CrossRef CAS PubMed.
- S. L. Mayo, J. Phys. Chem., 1990, 94, 8897 CrossRef CAS.
- M. Rehman, B. Y. Shekunov, P. York, D. Lechuga-Ballesteros, D. P. Miller, T. Tan and P. Colthorpe, Eur. J. Pharm. Sci., 2004, 22, 1–17 CrossRef CAS PubMed.
- I. M. Grimsey, M. Sunkersett, J. C. Osborn, P. York and R. C. Rowe, Int. J. Pharm., 1999, 191, 43–50 CrossRef CAS PubMed.
- S. C. Das, I. Larson, D. A. Morton and P. J. Stewart, Langmuir, 2010, 27, 521–523 CrossRef PubMed.
- F. Thielmann, D. J. Burnett and J. Y. Heng, Drug Dev. Ind. Pharm., 2007, 33, 1240–1253 CrossRef CAS PubMed.
- P. P. Ylä-Mäihäniemi, J. Y. Heng, F. Thielmann and D. R. Williams, Langmuir, 2008, 24, 9551–9557 CrossRef PubMed.
- J. Y. Heng, F. Thielmann and D. R. Williams, Pharm. Res., 2006, 23, 1918–1927 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and figures showing full synthon analysis for TBS, together with surface chemistry and the binding sites of polar probe on crystal surfaces of TBS and the experimental data from IGC measurements. See DOI: 10.1039/d0ce00026d |
| This journal is © The Royal Society of Chemistry 2020 |