
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Na2Ti3O7 nanorods by a V-assisted route and investigation of their battery performance
S.
Altin
*a,
S.
Demirel
b,
E.
Oz
a,
E.
Altin
c,
C.
Hetherington
d,
A.
Bayri
a and
S.
Avci
 *e
*e
aDepartment of Physics, Inonu University, Malatya, 44280, Turkey. E-mail: serdar.altin@inonu.edu.tr
bDepartment of Electricity and Energy, Igdir University, Igdir, 76000, Turkey
cIBTAM, Inonu University, Malatya, 44280, Turkey
dDepartment of Chemistry, Lund University, Lund, 22100, Sweden
eDepartment of Engineering Physics, Istanbul Medeniyet University, Istanbul, 34700, Turkey. E-mail: sevda.avci@medeniyet.edu.tr
First published on 25th February 2020
Abstract
We report the V-assisted synthesis of Na2Ti3O7 nanorods via a conventional solid state reaction technique. Energy dispersive X-ray spectroscopy (EDS) mapping showed that V-ions are not incorporated into the main structure of the nanorods but rather V behaves as a flux agent during the growth of the nanorods. The cyclic voltammetry (CV) analysis of the samples shows changes in the redox peaks as a function of V content. Our detailed ex situ X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM) and X-ray absorption spectroscopy (XAS) analyses after 1000 cycles show that the degradation mechanism is the formation of various titanium oxide impurity phases which inhibits the Na-ion diffusion.
1. Introduction
Although Li-ion batteries have conquered the portable electronics market, their limitations due to the high cost and availability of Li have prevented them to be used in large scale applications. Na is a promising alternative due to its natural abundance and similar intercalation properties to Li. Despite their rich redox chemistry, Na-ion batteries have lower energy density and voltage than Li-ion batteries. More studies have to be performed to optimize the electrode materials in the Na-ion battery field in order to catch up with the Li-ion battery technology. Most studies have been related to positive electrodes; however, less attention has been paid to the negative electrodes for Na-ion batteries.1,2The performance of a battery depends on the working potential which is the difference between the potentials of the positive and negative electrodes. Therefore, a low ionization voltage, i.e. between 0 and 2 V, is a good criterion for anode materials. Among the various types of anode materials for Na-ion batteries such as carbonaceous materials, Na alloys and compounds and Ti-based compounds,3 Na2Ti3O7 is promising due to its very low potential (0.3 V).4 Na2Ti3O7 consists of zigzag layers of TiO6 octahedra with a monoclinic crystal structure (P21/m space group). Up to 3.5 Na ions per formula unit can be intercalated into the interlayer space, leading to a theoretical capacity of 311 mA h g−1.5 Experimental capacity values are far from the theoretical value for now, therefore extensive research efforts are required to improve the capacity of Na2Ti3O7. Na2Ti3O7 can be synthesized via various techniques in several forms: nanosheet6 and nanorod7 forms via hydrothermal synthesis, bulk form from solid state reactions4 and microsphere form with a spray drying method.8 The capacity value of Na2Ti3O7 is very sensitive to its synthesis technique and its morphology. For instance, a capacity value of 188 mA h g−1 at a current rate of 0.1 C was reached by solid state synthesis.9 However, the capacity decreases to 60 mA h g−1 for the 60th cycle. Man et al. have synthesized sheet-like Na2Ti3O7 materials by solid state synthesis with hollow sphere TiO2 as the Ti source. Electrochemical measurements show that an initial charge capacity of 191 mA h g−1 at a 0.1 C rate decreases to 101 mA h g−1 after 50 cycles.10 Ding et al. synthesized carbon-coated Na2Ti3O7via the solid state reaction method. They report an initial discharge capacity of 215 mA h g−1 at a 20 mA current rate which shows a sharp decrease for the 25th cycle down to ∼60 mA h g−1.11 All these solid-state synthesis reports yield Na2Ti3O7 in bulk form. However, the effects of nanosized electrode materials on battery performance have been reported repeatedly12–14 and conventional solid-state synthesis is useful for large scale production due to its simplicity. Therefore, it is important to synthesize Na2Ti3O7 in nanosize via solid state synthesis. There has been one recent study reporting synthesis of a mixture of Na2Ti3O7/Na2Ti6O13 nanorods via the solid state reaction by adding carbon during synthesis.15 These nanorods showed promising electrochemical performance especially at high rates (%95Na2Ti3O7/%5Na2Ti6O13 with an initial specific discharge capacity of 92 mA h g−1 at a 5 C rate). In this work we report a new route for solid state synthesis of Na2Ti3O7 nanorods by adding vanadium. Our results confirm that the V ions do not diffuse into the lattice of Na2Ti3O7 nanorods, instead they behave as a flux agent in the main matrix that assists the formation of the nanorods. We have tested the electrochemical properties of Na2Ti3O7 nanorods in Na-ion batteries and used ex situ characterization techniques to study their capacity fade mechanism.
2. Experimental section
Nominal Na2Ti3−xVxO7 compounds, where x = 0.0, 0.025, 0.05, 0.1, and 0.15, were synthesized via the solid state reaction method. Powders of 99% purity Na2CO3, TiO2, and V2O5 were used as starting materials. The mixed powder was heated at 800 °C in air for 16 hours to remove the carbonates. After furnace cooling, the powder was mixed with an agate mortar and kept at 800 °C for 12 hours to obtain the final compositions.X-ray diffraction (XRD) measurements were performed using CuKα (λ = 1.5405 Å) radiation with a Siemens Bruker D5000 computer-controlled X-ray diffractometer at the University of Illinois at the Urbana-Champaign Materials Research Laboratory. The data were collected at a constant scanning rate of 2° min−1 at 2θ = 2–80°. Unit cell parameters were determined by the Rietveld refinement technique using the comprehensive General Structure Analysis System II (GSAS II) software.16
For the surface morphology analysis, a JEOL 6060LV general purpose scanning electron microscope (SEM) was used at the University of Illinois at the Urbana-Champaign Materials Research Laboratory. For the transmission electron microscopy (TEM) analysis, a JEOL 3000F with an energy dispersive X-ray spectroscopy (EDS) attachment was used at 300 kV at Lund University, Sweden. X-ray absorption fine structure (XAFS) analysis of the samples was performed at beamline 64 at DESY synchrotron in Germany.
Pure Na metal was used as a counter electrode for electrochemical characterization. The nominal Na2Ti3−xVxO7 compositions where x = 0, 0.025, 0.05, 0.1, and 0.15 were mixed with polyvinylidene fluoride (PVDF) and carbon black (CB) with a ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15 for CR2032 coin cells. The coin cells were pressed in an Ar gas filled glove box. Charge–discharge and cyclic voltammetry measurements were performed using a Bio-logic VPM3. Charge–discharge measurements were conducted at 0.2, 0.5, 1 and 2 C rates in the 0.5–2 V range. CV measurements were taken at 0.1 mV s−1 in the 0.1–2 V range.
:15:15 for CR2032 coin cells. The coin cells were pressed in an Ar gas filled glove box. Charge–discharge and cyclic voltammetry measurements were performed using a Bio-logic VPM3. Charge–discharge measurements were conducted at 0.2, 0.5, 1 and 2 C rates in the 0.5–2 V range. CV measurements were taken at 0.1 mV s−1 in the 0.1–2 V range.
3. Results and discussion
3.1. Growth of the nanorods
The growth of nano-size rod-like crystals is important in electrochemical processes due to their high surface area and short diffusion distances.17,18 Also, a crystalline structure is essential since ion diffusion occurs through certain crystallographic directions and planes.19 Although Na2Ti3O7 has been synthesized in nanorod form via various chemical techniques, solid state synthesis yields bulk materials for this compound.20,21 Sauvet et al. have reported the single phase formation temperature of Na2Ti3O7 to be 800 °C based on thermogravimetric and thermal differential analysis data.22Fig. 1(a) shows the XRD data of nominal Na2Ti3−xVxO7 compositions annealed at 850 °C. All the samples have the Na2Ti3O7 phase as the main phase. Fig. 1(b) shows the Rietveld refinement of the Na2Ti3O7 structure for the x = 0.1 sample. | ||
| Fig. 1 (a) XRD data of Na2Ti3−xVxO7 samples with nominal compositions where x = 0.00, 0.025, 0.05, 0.10 and 0.15. (b) Rietveld refinement of the Na2Ti3O7 structure for the nanorods scraped from the surface of the main matrix of the x = 0.1 sample. | ||
We have used the XRD data to calculate the lattice parameters and unit cell volume of the samples and observed that addition of V does not cause a significant change in the cell parameters, as listed in Table 1. This can also be confirmed by the XRD data shown in Fig. 1(a) where the peaks do not shift noticeably upon V addition. One possible explanation is the very small difference between the ionic radii of Ti4+ and V4+ ions (for 6-coordinations, the ionic radii are 74.5 pm and 72 pm, respectively). Another possible explanation is that the V ions do not incorporate with the lattice of Na2Ti3O7, instead they form a vanadium-containing impurity phase. We have detected impurity peaks in our XRD data; however, we could not identify these peaks due to their low intensity. These impurity peaks are not present in the samples with low V content (x = 0.025) and start appearing at the x = 0.05 sample. We have performed EDS analysis to observe if V is present in the nanorods as discussed later in this section.
| Sample | a (Å) | b (Å) | c (Å) | Volume (Å3) |
|---|---|---|---|---|
| 0.00 | 8.5685(14) | 3.80054(22) | 9.1252(15) | 291.125(41) |
| 0.025 | 8.5613(11) | 3.80255(15) | 9.1274(16) | 291.066(26) |
| 0.05 | 8.5619(11) | 3.80204(12) | 9.1264(11) | 291.026(15) |
| 0.10 | 8.5652(16) | 3.80156(17) | 9.1208(16) | 290.945(22) |
| 0.15 | 8.5644(13) | 3.80129(15) | 9.1252(14) | 291.017(19) |
Growth of crystals is usually initiated by a nucleus which is formed on the bulk part of the sample. Grain shapes of materials are directly related to the thermodynamic conditions of the environment such as temperature, pressure, internal energy, etc. A triggering mechanism is needed for the formation of rod-like grains since the rod-like grain formation requires more energy than that of the flake-like grains due to their high surface area.23 In our case, addition of V triggers the formation of Na2Ti3O7 nanorods. Fig. 2 shows the SEM images of nominal Na2Ti3−xVxO7 compounds. While the V-free sample (x = 0.0) does not contain any nanorods, even addition of a tiny amount of V (x = 0.025) activates the formation of nanorods.
 | ||
| Fig. 2 SEM images of Na2Ti3−xVxO7 samples where (a) x = 0, (b) x = 0.025, (c) x = 0.05, (d) x = 0.1 and (e) x = 0.15. | ||
Fig. 3 shows the TEM images of a nanorod and its diffraction pattern. The nanorod shows the single crystalline structure with growth along the b-axis. Values of the interplanar spacings were measured from the high resolution image and the obtained values (0.83 nm and 0.39 nm) correspond within error to the values calculated from the XRD data (Table 1).
 | ||
| Fig. 3 TEM images of a nanorod synthesized by V-substitution at the x = 0.05 level and before cycling. Analysis of the high resolution image and diffraction patterns taken from the same area give spacings along the b-direction as 0.39 nm and along the a-direction as 0.83 nm. | ||
The growth of one-dimensional crystals in the form of nanorods is a curious subject in materials science. There are several suggested models for the growth of these nanorods such as vapor–solid–liquid mechanisms, defect-assisted growth, etc.24–26 Based on our results, the most appropriate mechanism to explain the growth of Na2Ti3O7 nanorods is the defect-assisted growth. According to this model, incorporation of V in the lattice forms a defect and such defects on the surface of the main matrix causes formation of nuclei for the grains. The grains start to grow on the nuclei and the V defect does not diffuse into the structure of the nanorods; rather it behaves as a catalytic role in the growth process.27 To test this hypothesis we have performed EDS analysis to observe if the V-ions are present in the nanorods. We examined several nanorods and found that V-ions do not incorporate the structure of the nanorods. Fig. 4 shows investigations into one nanorod as an example. The STEM image was used to define a region for recording EDS spectra and mapping the main elements Na, Ti and O. The sum spectra were obtained from the mapping data and used to calculate atomic percentages. These were Na 16%, Ti 16%, O 68% and V 0.1%. The discrepancy between these numbers and the formula Na2Ti3O7 probably has two causes. One is that the calculated percentages depend on the value of thickness incorporated into the calculation; we estimated 200 nm but over the area of the map there is a large variation and hence uncertainty. The second is that on other occasions where spot analyses were attempted, we observed material loss at the site of the electron probe. The percentage for V was given at 0.1%. Values below around 1% mean that one normally concludes that an element is below the detection limit. The analysis of this EDS spectrum is complicated by an overlap of the V Kα peak at 4.95 keV and the Ti Kβ peak at 4.93 keV. The strong Ti Kα signal at 4.51 keV and the weaker Ti Kβ peak are clearly visible. The locations of V K peaks, where they should be present, are marked in red in Fig. 4. The, V Kβ peak location at 5.43 keV must be examined instead of the main V Kα peak. At 5.43 keV, there was no sign of a peak which allows us to say that V is not detected. We conclude that V-ions are not incorporated but instead play a catalytic role in the nanorod growth similar to the growth of Na0.44MnO2 nanorods as discussed in detail in our previous report.28
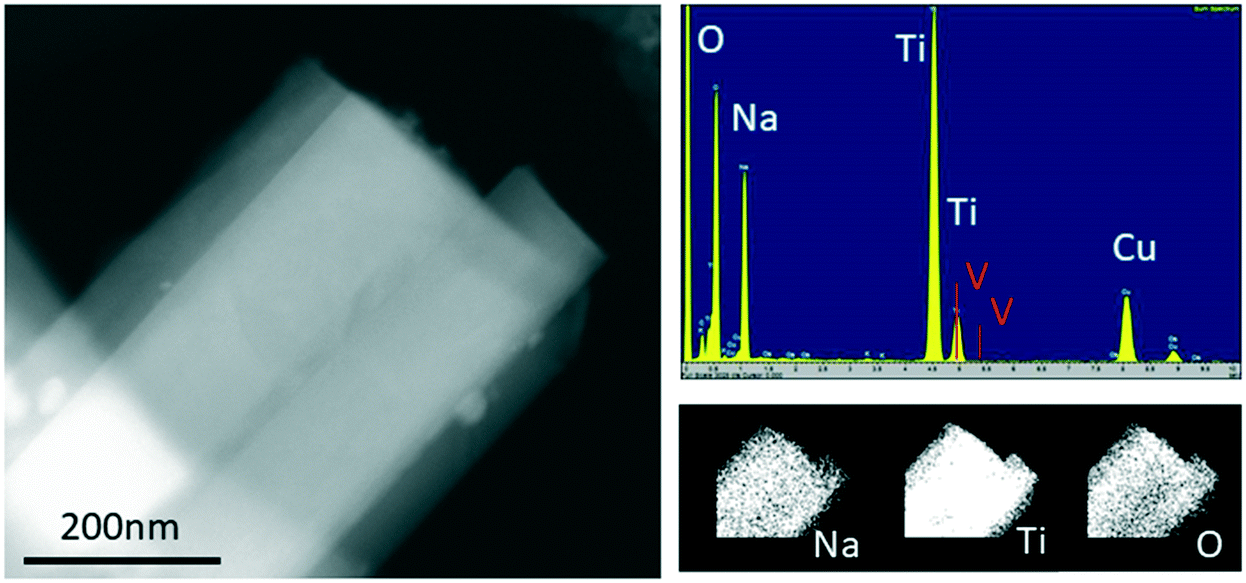 | ||
| Fig. 4 TEM-EDS image of a nanorod of the nominal Na2Ti3−xVxO7 composition where x = 0.05. On the left is a STEM dark field image. Upper right: The sum spectra recorded during mapping; peaks for O Kα, Na Kα, and Ti Kα are visible (Cu comes from the copper grid used as a sample holder). Lower right: The elemental maps indicate a uniform composition across the nanorod. | ||
Fig. 5(a) shows the Ti–K edge XANES spectra of the samples of the nominal NaVxTi3−xO2 compounds where x = 0.00–0.25. The XANES data show seven different featured peaks or kinks as labelled in Fig. 5(a). A. Niltharach et al. stated that A1 is related to the quadrupole transition from 1s to the t2g level of the TiO6 octahedron, A2 and A3 can be explained by the transition from 1s to 3d orbitals and B is attributed to the transition from 1s to 4p orbitals (detailed information can be found in ref. 29 and 30).
 | ||
| Fig. 5 (a) XANES data of nominal Na2Ti3−xVxO7 samples where x = 0.0, 0.025, 0.05, 0.1 and 0.15, (b) crystal structure of Na2Ti3O7, (c) FT XANES data of Na2Ti3−xVxO7 and (d) peak positions and sizes (ΔP = Pend − Pstart) obtained from the FT data depending on the V content. | ||
The crystal structure of Na2Ti3O7, obtained from Rietveld refinement, shown in Fig. 5(b), consists of three Ti sites, seven oxygen sites and two Na sites. Hennig et al. have reported that bond lengths obtained from the XRD data can be less reliable than the values obtained from the EXAFS data.31 We have used the Fourier transform (FT) EXAFS data to confirm the interatomic parameters (bond lengths) obtained from the XRD data. In the FT EXAFS data (Fig. 5(c)), the peaks are labelled P1, P2, P3, P4 and P5 (Fig. 5(d)) and corresponding bond types (listed in Table 2) are gathered from the XRD data. Each peak of the FT data consists of several bonds in the structure. Although the peak values of the FT EXAFS data provide the bond lengths of ions, the peak widths explain the statistical distribution of the bond lengths.
| Label in the FT data | Peak start (Å) | Peak end (Å) | Peak value (Å) | Bond types |
|---|---|---|---|---|
| P1 | 0.54 | 1.98 | 1.15 | Ti1–O1, Ti1–O2, Ti2–O2, Ti2–O3, Ti3–O4, Ti1–O5, Ti2–O6, Ti3–O1 |
| P2 | 2.02 | 3.21 | 2.6 | Ti2–Ti3, Ti2–O4, Ti3–O7, Ti2–O5, Ti1–O6, Ti1–O7 |
| P3 | 3.25 | 3.87 | 3.45 | Ti3–O6, Ti2–O7, Ti1–Ti2, Ti1–Na1, Ti2–Na1 |
| P4 | 3.90 | 4.56 | 4.2 | Ti2–O1, Ti3–O3, Ti1–Na2, Ti1–Na2, Ti3–Na2, Ti3–O2, Ti1–O3 |
| P5 | 4.63 | 5.93 | 5.05 | Ti1–O4, Ti3–Na1, Ti1–Ti3 |
3.2. Battery performance
Fig. 6a–e show the plot of the galvanostatic test profiles of nominal Na2Ti3−xVxO7 compounds up to 1000 cycles at a 0.5 C rate. The pristine compound, Na2Ti3O7, shows similar battery performance to that reported in the literature.32 A significant decrease in the capacity values upon increasing the cycle number is observed for most of the samples including the pristine compound. The lowest capacity fade after 1000 cycles is obtained for the x = 0.1 sample. | ||
| Fig. 6 Capacity–voltage graphs of nominal Na2Ti3−xVxO7 compositions (x = 0, 0.025, 0.05, 0.1 and 0.15). | ||
Fig. 7 shows the capacity change with increasing charge/discharge cycle number for Na2Ti3−xVxO7 up to 1000 cycles. Although the V substitution has a negative effect on the capacity of Na2Ti3O7, this material has a very good capacity retention property since its capacity remains almost constant between 300 and 1000th cycles. Even with the addition of vanadium, the crystal structure of the materials is stable upon charging/discharging. We have used both the main matrix and the nanorods to fabricate the electrodes. Fig. 7 shows that the pristine sample has 110 mA h g−1 and 42 mA h g−1 for the 1st and 1000th cycles, respectively.
 | ||
| Fig. 7 Capacity change with cycle number of nominal Na2Ti3−xVxO7 compositions (x = 0, 0.025, 0.05, 0.1 and 0.15). | ||
3.3. Degradation mechanism
The crystal structure of the active materials plays a crucial role in the battery performance. Investigations of the structural deformation during charge/discharge are expected to reveal the structural factors behind the capacity fade. Insertion/removal of the Na ions from the active materials causes changes in the crystal structure. Upon multiple repetitions of this process, these structural modifications become irreversible and detrimental for the battery life.The ex situ XRD data of the electrode materials after 1000 cycles as presented in Fig. 8 reveal impurity phases such as TiO, TiO2 and Ti7O13.
 | ||
| Fig. 8 Ex situ XRD data of nominal Na2Ti3−xVxO7 (x = 0, 0.025, 0.05, 0.1 and 0.15) compounds after 1000 cycles. | ||
We also performed ex situ XAS measurements after the 1st, 25th, 50th, 100th and 500th cycles for the x = 0.10 sample to determine the major structural changes upon repeated cycling as seen in Fig. 9. We have observed 6 different peaks in the XAFS data as observed in uncycled samples (Fig. 5(a)). C1, C2 and C3 peaks have relatively higher intensity than those of the uncycled samples and the cycling process changes the spectrum slightly. The change in the intensity of the C-signed peaks in the XANES data is related to formation of TiO2, TiO and Ti7O13 phases since the environment of Ti ions change with the formation of these phases. In addition, the FT data show a similar behavior with the uncycled sample. It is clear that the main phase remains to be present upon cycling and the bond lengths of impurity phases as shown in the XRD graph (Fig. 8) should be the same length in the existing peaks of the main phase as seen in Fig. 9(b) and (c).
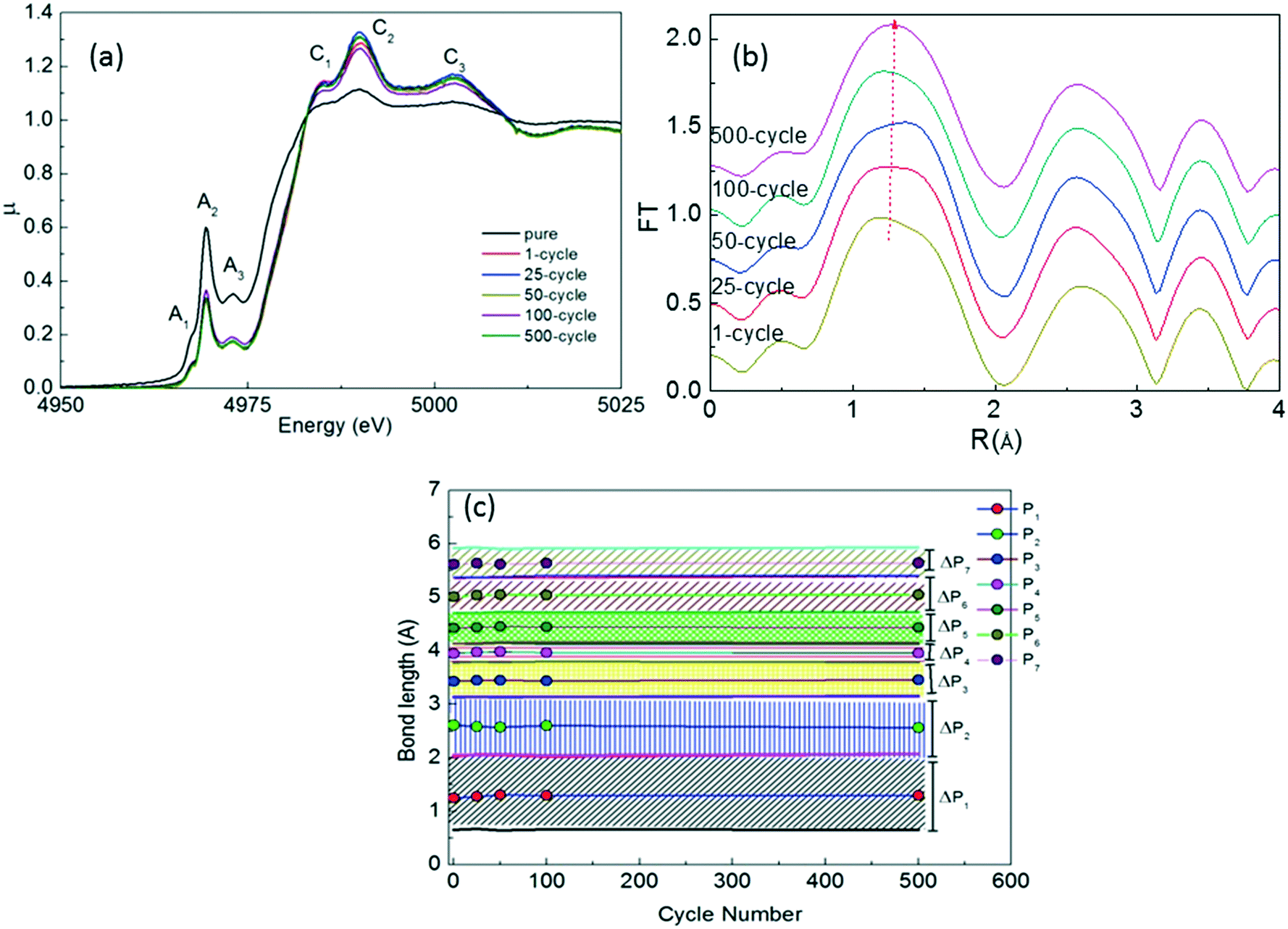 | ||
| Fig. 9 (a) XANES spectra, (b) FT graphs and (c) peak positions and ΔP values (in FT graphs) of x = 0.10 Na2Ti3O7 after 500 cycles. | ||
TEM was used to look at the nature of structural changes that occur during cycling. In Fig. 10 we show the TEM images of a nanorod of the x = 0.10 sample ‘before’ and ‘after’ 1000 cycles. The ‘before’ sample has a clean overall appearance (top left), and the high resolution image (top right) has a uniform crystalline appearance up to the surface. The ‘after’ sample (bottom left) shows variations in intensity across its width and there is an extra material attached to the surfaces. The high resolution image (bottom right) shows loss of crystallinity. The interplanar spacing measured in the ‘before’ material was 0.82 nm, the same within error as reported in Fig. 3. In the ‘after’ material, the spacing lies in the range of 0.68 to 0.70 nm. This clearly shows the structural deformation upon repeated cycling.
 | ||
| Fig. 10 TEM images of a nanorod of the x = 0.10 sample before cycling and after 1000 cycles. | ||
Conclusions
In this work we report the solid state synthesis of Na2Ti3O7 nanorods via a defect assisted route upon V addition. Among the nominal Na2Ti3−xVxO7 compositions where x = 0.0, 0.025, 0.05, 0.1 and 0.15, we observed that the pristine sample (x = 0.0) has the bulk form while addition of even a small amount of V in the nominal composition causes formation of nanorods. Our TEM-EDS analysis shows that the nanorods do not contain any vanadium. We attribute the growth of the nanorods to a defect assisted growth mechanism which confirms our claim regarding the catalytic role of vanadium in the nanorod growth. We have also tested battery performances of our samples. The x = 0.10 sample shows the best capacity retention among all the samples, therefore we have examined the capacity fade mechanism of this sample via ex situ XRD, XAS and TEM studies. We found that repeated cycling causes formation of various titanium oxide phases and significant morphological deformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Inonu University Research Council with contract number of BAP-2015/85. Dr. S. Demirel was supported by the TUBITAK 2214-A International scholarship program. Synchrotron and TEM studies were performed through funding from the EU-H2020 research and the innovation program under grant agreement No. 654360 having benefitted from the access provided by DESY in Germany and by Lund University in Sweden within the framework of the NFFA-Europe Transnational Access Activity.References
- V. L. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, 158, A1011 CrossRef CAS.
- Y. Liu, Z. Sun, K. Tan, K. D. Di, J. Sun, L. Liang, L. Hou and C. Yuan, J. Mater. Chem. A, 2019, 7, 4353–4382 RSC.
- C. Yang, Y. Liu, X. Sun, Y. Zhang, L. Hou, Q. Zhang and C. Yuan, Electrochim. Acta, 2018, 271, 165–172 CrossRef CAS.
- P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- J. Ni, S. Fu, C. Wu, Y. Zhao, J. Maier, Y. Yu and L. Li, Adv. Energy Mater., 2016, 6, 1502568 CrossRef.
- Z. Zhang, J. B. M. Goodall, S. Brown, L. Karlsson, R. J. H. Clark, J. L. Hutchison, I. U. Rehman and J. A. Darr, Dalton Trans., 2010, 39, 711–714 RSC.
- Y. V. Kolen'ko, K. A. Kovnir, A. I. Gavrilov, A. V. Garshev, J. Frantti, O. I. Lebedev, B. R. Churagulov, G. Van Tendeloo and M. Yoshimura, J. Phys. Chem. B, 2006, 110, 4030–4038 CrossRef PubMed.
- W. Zou, J. Li, Q. Deng, J. Xue, X. Dai, A. Zhou and J. Li, Solid State Ionics, 2014, 262, 192–196 CrossRef CAS.
- H. Pan, X. Lu, X. Yu, Y.-S. Hu, H. Li, X.-Q. Yang and L. Chen, Adv. Energy Mater., 2013, 3, 1186–1194 CrossRef CAS.
- M. Xie, K. Wang, R. Chen, L. Li and F. Wu, Chem. Res. Chin. Univ., 2015, 31, 443–446 CrossRef CAS.
- C. Ding, T. Nohira and R. Hagiwara, J. Power Sources, 2017, 354, 10–15 CrossRef CAS.
- C. Jiang, E. Hosono and H. Zhou, Nano Today, 2006, 1, 28–33 CrossRef.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- C.-K. Ho, C.-Y. V. Li and K.-Y. Chan, Ind. Eng. Chem. Res., 2016, 55, 10065–10072 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, IUCr, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294 RSC.
- T. Kawamura, M. Makidera, S. Okada, K. Koga, N. Miura and J. Yamaki, J. Power Sources, 2005, 146, 27–32 CrossRef CAS.
- M. Xu, Y. Niu, C. Chen, J. Song, S. Bao and C. M. Li, RSC Adv., 2014, 4, 38140–38143 RSC.
- P. Slamet, W. B. Widayatno, A. Subhan and B. Prihandoko, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 432, 012058 CrossRef.
- A. Rudola, K. Saravanan, C. W. Mason and P. Balaya, J. Mater. Chem. A, 2013, 1, 2653 RSC.
- A. L. Sauvet, S. Baliteau, C. Lopez and P. Fabry, J. Solid State Chem., 2004, 177, 4508–4515 CrossRef CAS.
- Crystal Growth - From Fundamentals to Technology, ed. G. Müller, J. J. Metois and P. Rudolph, Elsevier, 2004 Search PubMed.
- H. Yung-Jung and S.-Y. Lu, J. Phys. Chem. B, 2005, 106, 4398–4403 Search PubMed.
- H. K. Yu and J.-L. Lee, Sci. Rep., 2015, 4, 6589 CrossRef PubMed.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312 RSC.
- Y. S. Park, S.-H. Lee, J.-E. Oh, C.-M. Park and T.-W. Kang, J. Cryst. Growth, 2005, 282, 313–319 CrossRef CAS.
- S. Demirel, E. Oz, E. Altin, S. Altin, A. Bayri, P. Kaya, S. Turan and S. Avci, Mater. Charact., 2015, 105, 104–112 CrossRef CAS.
- A. Niltharach, S. Kityakarn, A. Worayingyong, J. Thienprasert, W. Klysubun, P. Songsiriritthigul and S. Limpijumnong, Phys. B, 2012, 407, 2915–2918 CrossRef CAS.
- F. Farges, G. E. Brown and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1809–1819 CrossRef CAS.
- C. Hennig, G. Reck, T. Reich, A. Roßberg, W. Kraus and J. Sieler, Z. Kristallogr., 2003, 218, 37–45 CAS.
- P. Li, W. Wang, S. Gong, F. Lv, H. Huang, M. Luo, Y. Yang, C. Yang, J. Zhou, C. Qian, B. Wang, Q. Wang and S. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 37974–37980 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |