
Syntheses, structures, and magnetic properties of three supramolecular isomeric Cu(ii) square grid networks: solvents effect on the ligand linkages†
 *b
*b
Abstract
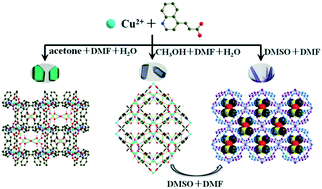
Reactions of deprotonated (E)-3-(quinolin-4-yl) acrylic acid (QCA) with Cu(NO3)2 in different solvents/templates gave three supramolecular isomers: [Cu(QCA)2]2·6H2O·(CH3)2CO (1), [Cu(QCA)2]·5H2O·CH3OH (2) and [Cu(QCA)2]·DMSO (3). Single-crystal X-ray diffraction analyses revealed that 1, 2 and 3 are all 2D square grid networks, but with different metal fragments and ligand conformations, based on the remarkable effect of solvents. Interestingly, a catenarian water pentamer and a water tape composed of cyclic water pentamers were observed in the crystal lattice of 1 and 2 respectively, which played a crucial role in not only the stabilization but also the template direction of the formation of the 3D stacking crystal host. Moreover, because of the different coordination modes and intramolecular interactions, 1, 2 and 3 showed different magnetic properties.
 Please wait while we load your content...
Please wait while we load your content...

