
Vacancy-engineered catalysts for water electrolysis
Songa
Choi†
 ,
Yeji
Park†
,
Heesu
Yang†
,
Haneul
Jin
,
Gracita M.
Tomboc
and
Kwangyeol
Lee
*
,
Yeji
Park†
,
Heesu
Yang†
,
Haneul
Jin
,
Gracita M.
Tomboc
and
Kwangyeol
Lee
*
Department of Chemistry and Research Institute for National Sciences, Korea University, Seoul 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
First published on 7th January 2020
Abstract
The development of electrochemical energy conversion and storage technologies is pivotal to the full-fledged utilization of renewable energy sources. The successful commercial application of water electrolysis to produce hydrogen gas, in particular, requires highly active electrocatalysts that can operate for prolonged periods. However, the high activity and high durability of electrocatalysts are often mutually exclusive. Recent studies have demonstrated that vacancy engineering might effectively modulate the electronic structures of catalysts, which can lead to high catalytic activity. Furthermore, it has been shown that vacancies are closely related to catalyst stability under operational conditions. To understand the benefits of vacancies in the catalyst structures, we discuss the recent advances in the development of vacancy-engineered catalysts for water electrolysis. In addition, we discuss the present limitations in this nascent field and provide directions for valuable future research.
 Haneul Jin | Dr. Haneul Jin (Front line), Heesu Yang, Songa Choi, Yeji Park, and Dr. Gracita M. Tomboc (back line: from left to right) are currently working in inorganic chemistry under the supervision of Prof. Kwangyeol Lee at the Department of Chemistry, Korea University (South Korea). H. Yang has been working on synthesis of nanoscale materials and their applications toward water splitting reaction. S. Choi is involved in designing noble metal-based functional nanomaterials for energy conversion/storage and CO2 conversion. Y. Park is interested in the design of hetero-nanostructured catalysts and the development of functional nanomaterials for energy conversion. Dr. G. M. Tomboc is focused on advanced materials processing of ideal electrode materials for electrochemical energy storage system applications such as supercapacitor and water splitting process. Dr. H. Jin is interested in the development of synthetic methods for phase-controlled nano-catalysts and investigation of water splitting reaction mechanism on the heterogeneous catalyst surfaces. |
 Kwangyeol Lee | Professor Kwangyeol Lee (born in 1971) obtained his Ph.D. degree (1997) in Chemistry from the University of Illinois at Urbana–Champaign. After fulfilling his military obligation, he joined Korea University in 2003 as a Chemistry faculty member, before being appointed professor. He is the recipient of the 2009 Wiley-KCS Young Scholar Award and 2019 Excellent Research Award (KCS Inorganic Chemistry Division). His current interests lie in the development of synthesis methods for nanoscale materials, applications of nanomaterials in biomedical fields, and development of nanotechnologies to support the environment by creating sustainable energy sources. |
1. Introduction
With the rapid consumption of fossil fuels and the associated serious environmental problems, the global demand for sustainable energy production from carbon-neutral fuels has increased continuously in the past decades. The production of hydrogen via electrochemical water splitting is considered one of the most promising energy sources due to the high energy density of hydrogen and the pollutant-free production process. However, the large operational potential window for the main water splitting reactions—hydrogen evolution reaction (HER) and oxygen evolution reaction (OER)—significantly hinders the commercialization of the water electrolysis system. Catalysts play a vital role in efficient energy conversions by decreasing the electrical energy usage during water electrolysis. Therefore, the development of active and durable catalytic materials for both HER and OER is an essential requirement for the advancement of water electrolysis technology.Efficient catalysts require optimized surface physicochemical properties for facile adsorption and desorption of intermediates. Recently, vacancy engineering on the catalyst surfaces has emerged as an effective strategy to enhance the HER and OER performances by controlling the surface electronic and atomic structures. The modification of the surface electronic structure adjusts the adsorption energy of the intermediates (e.g., *H for HER; *OH, *OOH for OER) on the catalyst surface and promotes charge transfer at the interface between the electrolyte and the electrodes.1,2 The coordination-unsaturated structure derived from vacancy engineering generates catalytically active sites for water electrolysis on the catalyst surface. However, the development of vacancy-engineered catalysts for electrochemical water splitting is only in its infancy.
In this review, we provide a brief overview of the vacancy engineering of electrocatalysts for water-splitting reactions, focusing on the nature of the vacant sites on the catalysts and their effect on the water-splitting reaction. First, we describe the three aspects of vacancy engineering that affect catalytic performances: (1) formation of low-coordination sites, (2) reaction kinetics control, and (3) modulation of electronic structures. Thereafter, we summarize the performance of vacancy-engineered catalysts in their applications toward HER and OER. Finally, we discuss the current limitations of vacancy-engineered catalysts and the future research directions for the development of high-performance vacancy-engineered catalysts for the electrochemical water-splitting process.
2. The effects of catalyst vacancies on electrochemical water splitting
The formation of the vacancies on an electrocatalyst can effectively modulate the local atomic structure, which creates a low-coordinated local geometry, and changes the electron configuration, thereby leading to an increase in the number of active sites, surface energy, and electron transfer, which are crucial for the efficient performance of catalysts in electrochemical water splitting.3 Therefore, understanding the relationship between the vacancy and electrocatalytic performance is important for the rational design of a high-performance catalyst toward electrochemical HER and OER (Scheme 1). In this section, we discuss the beneficial effects of vacant sites on the electrocatalytic performance toward water splitting.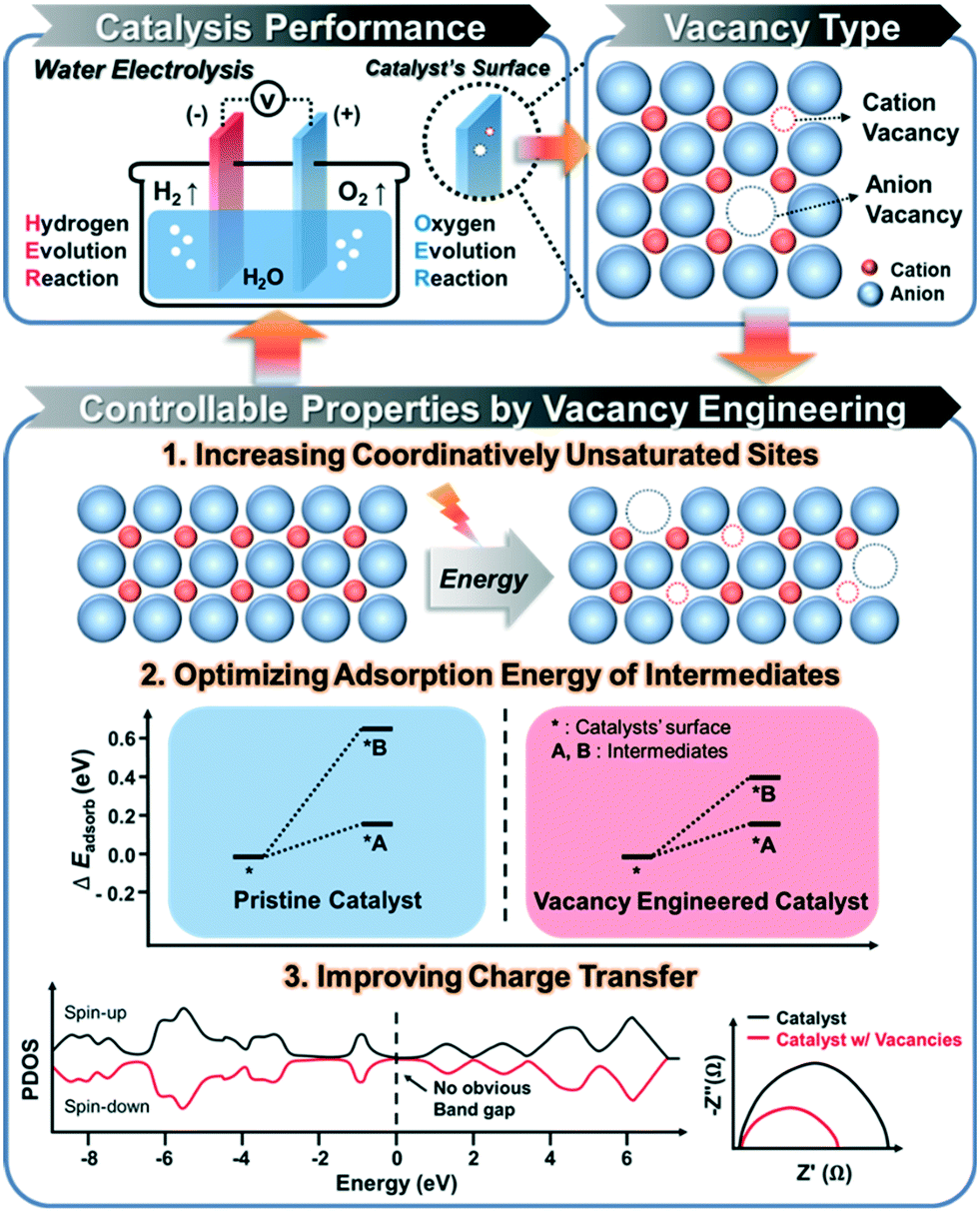 | ||
| Scheme 1 Illustration of the strategies for enhancing the performance of electrocatalytic water splitting via vacancy engineering. | ||
2.1. Formation of low-coordination sites
Low-coordinated sites serve as active sites on catalysts to promote catalytic reactions4 owing to the high density of their dangling bonds, namely coordinatively unsaturated sites (CUSs), which are highly reactive. For instance, micro-kinetics mode-based density-functional theory (DFT) calculations demonstrated that low-coordinated gold atoms at the corners and edges of nanoparticles act as active sites in a CO oxidation reaction.5 Similarly, vacant sites can lower the coordination number (CN) of the adjacent sites and modulate the local electronic structures. Sun et al. demonstrated that the ultrathin Co3O4 structure offers a considerably large fraction of coordinatively unsaturated surface atoms that serve as active sites to facilitate the OER.6 As shown in Fig. 1A, the adsorption energy of H2O molecules on Co3+ atoms was optimized by the gradual decrease in their CNs from 6 to 3, thereby indicating that a low CN could lead to high catalytic activity for the OER. For porous Co3O4 thin sheets, the presence of numerous pores decreased the CN of Co3+ atoms to 4 or even up to 3, and the thickness of half a unit cell enabled them to expose all the Co3+ atoms on their surfaces, thereby demonstrating that all the Co3+ atoms could function as active sites to catalyze the water oxidation reactions. Furthermore, the CUSs formed by S-vacancies on the edges of MoS2 were demonstrated as active catalytic sites and were considered key descriptors to evaluate the catalytic activity of MoS2-based materials.7–9 When the S-vacancies are introduced, new bands appear in the gap near the Fermi level, thereby indicating that these states are localized around the S-vacancy (Fig. 1B). Additionally, the bands move closer to the Fermi level and increase the number of gap states with increasing S-vacancies (Fig. 1C), thereby leading to an increase in the active site density and improvement of the HER catalytic performance. Furthermore, the electronic structure of MoS2 was highly dependent on the coordination of the Mo metal and its d-electrons. A semiconducting behavior and metallic conductivity can be observed when the non-bonding d-orbitals are fully and partially occupied, respectively.10 In addition, the high exposure of the edges of MoS2 nanosheets with unsaturated coordination and dangling bonds is crucial for their application in catalysis, electronics, and sensors, which require the involvement of a highly active surface.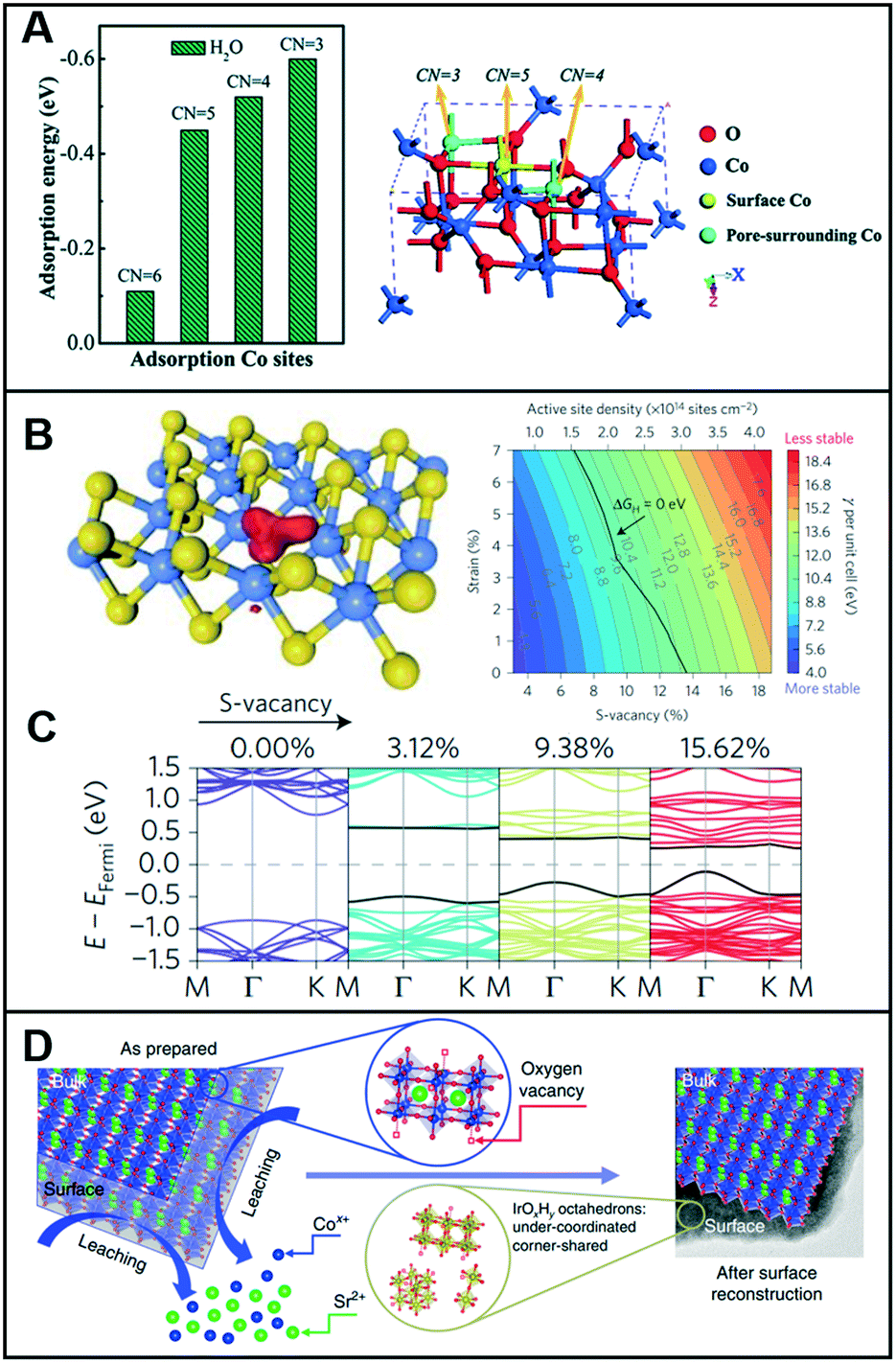 | ||
| Fig. 1 (A) Calculated adsorption energy for the H2O molecules on Co3+ sites with different CNs (left), and crystal structure showing the different CNs for surface and pore-surrounding Co3+ atoms. Reproduced with permission from ref. 6. Copyright 2014 Royal Society of Chemistry. (B) Kohn–Sham orbitals (red) are localized around the S-vacancies in MoS2 (left) and colored contour plot of surface energy per unit cell γ (with respect to the bulk MoS2) as a function of the S-vacancy and uniaxial strain (right). Color bar represents the value of γ. (C) Band structure with increasing S-vacancy concentrations (calculated using the full 4 × 4 unit cell). Reproduced with permission from ref. 9. Copyright 2016 Nature Publishing Group. (D) Schematic illustrating the surface reconstruction over the SrCo0.9Ir0.1O3−δ surface. Reproduced with permission from ref. 11. Copyright 2019 Nature Publishing Group. | ||
Chen et al. designed a pseudo-cubic SrCo0.9Ir0.1O3−δ perovskite that exhibits in situ formed vacancies during electrochemical cycling.11 During the electrochemical treatment within the potential range of 1.0–1.7 V at a scan rate of 10 mV s−1 in 0.1 M HClO4, Sr and Co were leached out, and a surface reconstruction occurred, as shown in Fig. 1D. Then, the SrCo0.9Ir0.1O3−δ perovskites contained a higher number of oxygen vacancies in the lattice than in its initial pseudo-cubic structure. X-ray absorption spectroscopy (XAS) analysis revealed that compared to the case in IrO2, the valence state of Ir in SrCo0.9Ir0.1O3−δ slightly shifted toward >4+. Assuming that the valence state of Ir is 5+, the δ (oxygen non-stoichiometry) in SrCo0.9Ir0.1O3−δ can reach a minimum value, which indicates that a high number of oxygen vacancies must exist in SrCo0.9Ir0.1O3−δ and that both Co and Ir must be highly under-coordinated. To support this assumption, Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra at the Ir LIII-edge were recorded to demonstrate that the Ir in SrCo0.9Ir0.1O3−δ is highly under-coordinated with a CN of ∼4.9 and that the under-coordinated IrOx octahedron considerably improved the catalytic activity toward the OER.
2.2. Optimizing the adsorption energy of intermediates
Both HER and OER mechanisms consist of multi-step reactions such as adsorption, reduction/oxidation, and desorption processes;12,13 their catalytic surfaces require suitable surface energies at each step.14 Luo et al. showed that the vacancy-induced hydrogen binding energy optimization in mesoporous MoO3−x could exhibit excellent HER activity under both acidic and alkaline conditions.15 DFT calculation revealed that the oxygen vacancies close to Mo5+ served as electron acceptors and favored the adsorption of water molecules in alkaline media (or H3O+ in acidic media), which lowered the HER energy barrier. The HER process was facilitated by the cooperation between Mo5+ and oxygen vacancies that led to the promotion of H2O adsorption and H–H bond formation on two adjacently adsorbed Hads and H2Oads species, as shown in Fig. 2A. Under an alkaline condition, a fast charge-transfer process was the only effective method of obtaining OH− during the reduction and formation of adsorbed H2Oads and Hads species and the release of H2. Additionally, Mo5+ ions donate electrons to become Mo6+ and then accept electrons to transform back to Mo5+. Similarly, Cheng et al. demonstrated that 2D Cr2CO2, modified with a transition metal and with engineered carbon vacancy, could improve the HER performance.16 The formation of carbon vacancies (VC) in 2D Cr2CO2 can be a natural phenomenon because the calculated formation energy of VC decreased as the percentage of VC increased. Additionally, when the concentration of VC reached up to 25%, the ΔGH* of VC-Cr2CO2 was very close to the ΔGH* value obtained for the perfect CrCO2. This suggests that VC-Cr2CO2 could provide an optimal reaction kinetics toward HER.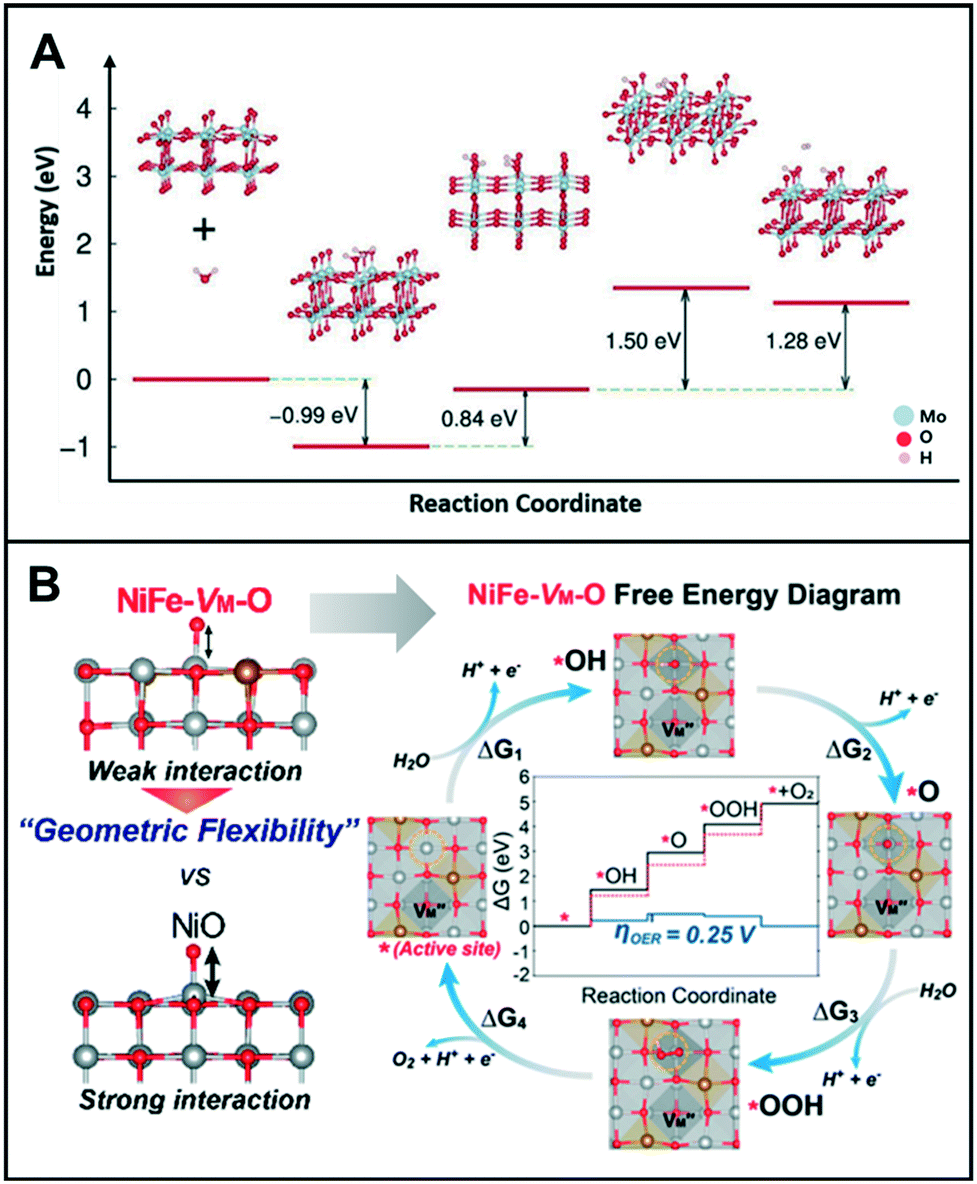 | ||
| Fig. 2 (A) Proposed reaction pathway, and the energy barrier profiles of mesoporous MoO3−x in 0.1 M KOH. Reproduced with permission from ref. 15. Copyright 2016 Wiley-VCH. (B) Free energy diagrams of NiFe-VM-O at 0 V (black line) and 1.23 V (blue line). Red dashed lines correspond to the free energies of the ideal catalyst with 0 V overpotential. Top views of each reaction step are presented. Each color indicates Ni (gray), Fe (brass yellow), and O (red). Reproduced with permission from ref. 22. Copyright 2019 American Chemical Society. | ||
On the contrary, it is imperative to control the reaction kinetics for the OER because of the multi-electrons (4e−) participating in this process.14,17,18 The rate-determining step (RDS) of the OER may vary depending on the O-adsorption/desorption on the catalyst surface. This implies that the formation of OOH is the RDS on a weak O-adsorbate and vice versa.12,19 Thus, the optimization of the bond strength between the intermediates and catalyst is crucial to facilitate the OER process. Additionally, the nucleophilic attack of adsorbed oxygen by water/hydroxide has been proposed to describe the mechanism of O–O bond formation during the OER. Pfeifer et al. revealed that the electrophilic oxygen species adsorbed on the surface and in the subsurface of amorphous Ir were indispensable for the high activity of hydrated amorphous IrIII/IV oxyhydroxide (IrOx), compared to crystalline IrO2.20 During the CO oxidation, they determined which oxygen atom in IrOx (OI− or OII−) may serve as the active species for the OER. They found that bulk OI− species are more electrophilic than their bulk OII− counterparts, as they were weakly bound to IrOx. The reactivity of the OI− species was suspected to play a critical role in the OER when reacted with OH/H2O to form the OOH intermediate because the electrophilic OI− species in amorphous IrIII/IV oxyhydroxide could be susceptible to nucleophilic attacks. In line with this, Nong et al. reported that Ni leaching during catalyst activation at high potential could lead to the formation of Ir lattice vacancies and numerous d-band holes in the formed IrOx shell in IrNi alloy nanoparticles.21 The oxygen ligands adjacent to the lattice vacancies were suggested to possess an electrophilic character that initiates the formation of O–O bonds and weakens the kinetic barrier during the OER, eventually resulting in high activity.
Meanwhile, Lee et al. investigated the transition metal (TM) vacancies (VM) formed during the simultaneous incorporation of Ni and Fe metals into the same crystal structure.22 The different preferential oxidation states (2+ for Ni and 3+ for Fe) would cause the VM to achieve charge neutrality with oxygen; the addition of Fe3+ into a NiO matrix resulted in the formation of NiFe-VM-O. As shown in Fig. 2B, this vacancy formation altered the bond strengths of adjacent Ni–O and Ni–TM, which improved the catalytic activity of the modified catalyst as compared to its original state, NiO. In terms of electronic structure modification of the active site, the energy of O-adsorption in NiFe-VM-O was lower than that in NiO owing to its flexible geometric distortions that can accommodate adsorbates easily.
2.3. Improving charge transfer
Most metal oxides have lower electrical conductivity than metals, resulting in lower electrochemical efficiency. Oxygen vacancy-engineered catalysts have been suggested to overcome the low electrical conductivity problem of transition metal oxides.23–25 Wang et al. reported that reduced mesoporous Co3O4 nanowires (NWs) with abundant oxygen vacancies have lower charge-transfer resistance than the pristine Co3O4 NWs.26 The total density of states (TDOSs) and the projected DOSs (PDOSs) calculation showed that the oxygen vacancies in Co3O4 NWs induced new defect states within the bandgap (Fig. 3A). The electron occupancy of the gap state was classified into three different states of occupied electrons, namely empty, occupied by one electron, or by two electrons, denoted as VO2+, VO+, VO, respectively. According to the calculated formation energy (Fig. 3B), VO2+ has the lowest formation energy for all the values of the Fermi level in the bandgap, indicating that the oxygen vacancy is most likely formed as VO2+. When the two electrons are excited, the formation energy of VO2+ is about 1 eV. These calculated values suggested that the reduced Co3O4 NWs can easily produce the oxygen vacancies with two positive charges (VO2+), and the two electrons are delocalized near the oxygen vacancy and are easily excited into the conduction band, thus leading to enhanced electrocatalytic performance. Likewise, other metal oxides such as NiO nanorods (NRs) also showed great HER activities owing to the vacancy-induced electronic structural modification.1 Zhao et al. reported the synthesis of ultrathin NiFe-layered double hydroxide (NiFe LDH-UF) nanosheets containing multi-vacancies, such as metal and oxygen vacancies, which enhanced their electronic conduction and HER reaction kinetics.27 DFT calculations revealed the effect of multi-vacancies (VO, VNi, and VFe) on the electronic structure of the NiFe LDH ultrafine nanosheets. While the monolayer and multi-layered LDH had noticeable band gaps, their calculated DOSs showed that there is no noticeable band gap around the Fermi level in the LDH-UF. This demonstrates the semi-metal-like character of the modified material, which can result in the enhancement of the electrical conductivity. Additionally, LDH-UF showed a lower charge-transfer resistance during electrochemical tests, compared with the LDH-bulk and LDH-monolayer, supporting the claimed excellent electrical conductivity.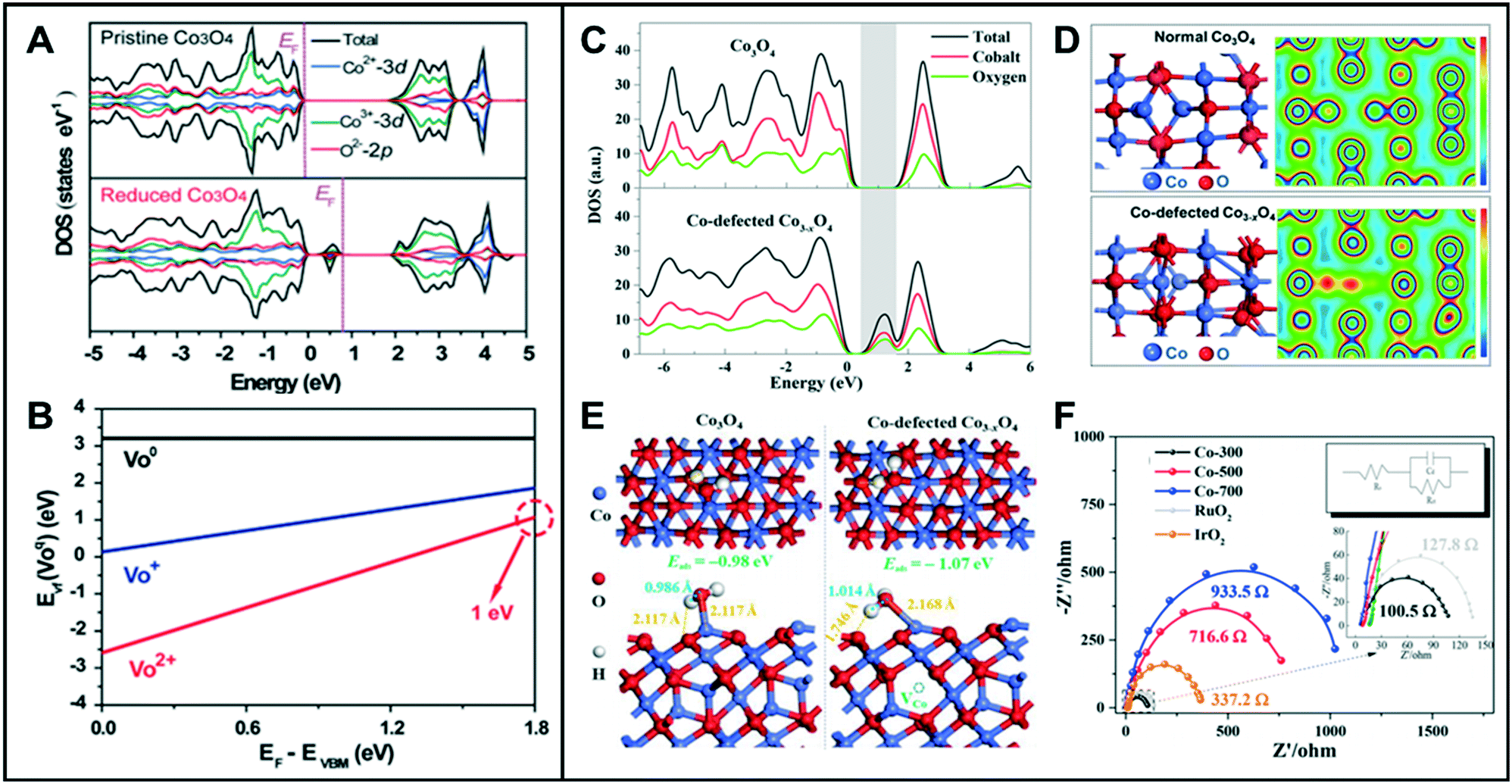 | ||
| Fig. 3 (A) TDOS and PDOS of pristine Co3O4 and reduced Co3O4 with oxygen vacancies. (B) Calculated formation energies for the reduced Co3O4. VO2+, VO+, and VO, denote that the gap state is empty, occupied by one electron, and occupied by two electrons, respectively. Reproduced with permission from ref. 26. Copyright 2014 Wiley-VCH. (C) TDOS and PDOS for Co3O4 and Co-defected Co3−xO4. (D) Optimized cell structures and the corresponding charge density mapping of normal Co3O4 and Co-defected Co3−xO4. (E) Optimized structures of water adsorbed at Co on the (111) surface (top and side views) of Co3O4 (left) and Co3−xO4 (right). (F) Electrochemical impedance spectroscopy (EIS) (inset: EIS fitting model; electrolyte resistance RS, charge-transfer resistance Rct, capacitive reactance Cd). Reproduced with permission from ref. 3. Copyright 2018 American Chemical Society. | ||
Similar to anion vacancies, cation vacancies can promote the charge transfer by modifying the electronic structures of the catalysts. Zhang et al. demonstrated that Co deficient Co3−xO4, with structural distortion and electronic delocalization, enhanced the rate of carrier transport to participate in OER.3 As the Co vacancies in Co3O4 are formed, the DOS for the occupied states from ca. 0.50 eV above the Fermi level increased and then showed a very small energy gap (Fig. 3C). The electronic structures of Co3−xO4 showed a considerable charge depletion, with more overlap areas of the electron wave-function with neighboring O atoms compared to those of the pristine Co3O4 (Fig. 3D). The charge density of Co3−xO4 increased around the conduction band edge to form a more dispersive electronic structure than that of pristine Co3O4, which resulted in a fast carrier transport for the OER. Moreover, the deformation charge densities of adsorbed H2O on Co3O4 and Co3−xO4 revealed that the Co-defective surface is more beneficial for the adsorption and desorption of water. When H2O molecules were adsorbed onto the surface of the Co3−xO4 catalyst, one H atom from the adsorbed water of Co3−xO4 showed relatively strong interaction with Os (surface O of Co3−xO4), which resulted in the extension of the H–OH bond length of the adsorbed water to 1.014 Å, compared to 0.986 Å for normal Co3O4. This result indicated that the Co-defective structure has the ability to facilitate the adsorption/desorption of intermediates and to improve the electronic conductivity, thus being highly favorable toward water splitting (Fig. 3E and F).
3. Classification of vacancy-engineered nanostructures and their catalytic performances in HER and OER
3.1. Anion vacancy
Among various forms of vacancies, anion vacancies are the most commonly used to improve catalytic performance because they are easily formed and can often generate an ideal Fermi level for water splitting reaction.9,26,28,29As mentioned in a previous section, low-coordination metal atoms are identified as the active sites of metal-oxide catalysts for water-splitting reaction.30 Bao et al. synthesized oxygen-vacancy-rich NiCo nanosheets (NiCo-r) and compared the results with bulk NiCo and oxygen-vacancy-poor NiCo nanosheets (NiCo-p).28 The O 1s X-ray photoelectron spectroscopy (XPS) spectra of the three materials exhibited a peak at 531.2 eV, with NiCo-r showing the highest intensity, which may be correlated to its low oxygen coordination and the high number of oxygen vacancies present compared to those in the others (Fig. 4A). The NiCo-r nanosheets showed enhanced activity toward the OER (320 mV at 10 mA cm−2) than oxygen-vacancy-poor or bulk catalysts. This result was consistent with DFT calculation, which revealed that NiCo2O4 with oxygen vacancies has lower adsorption energy for H2O (−0.75 eV) compared to the perfect surface (−0.41 eV) (Fig. 4B).
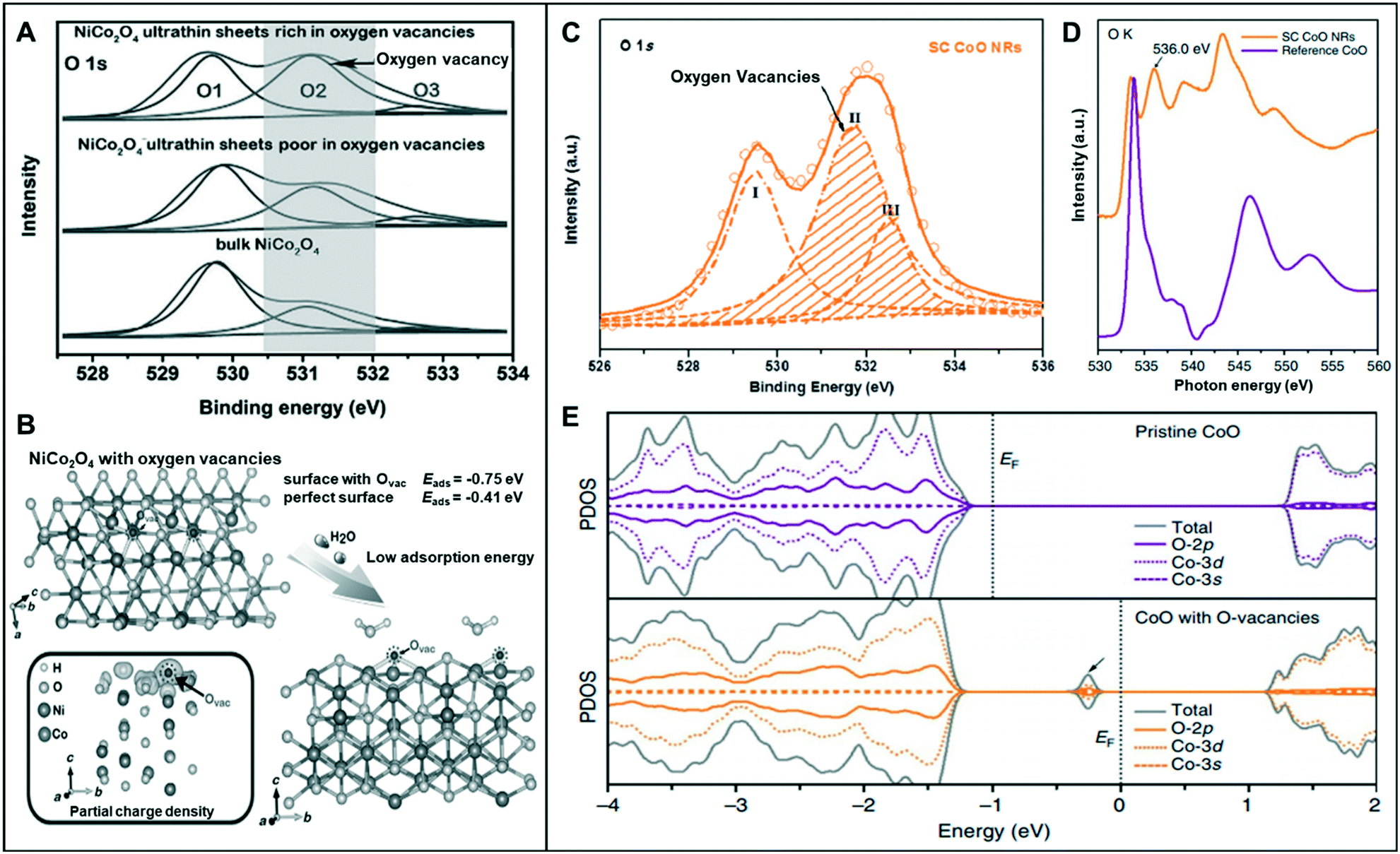 | ||
| Fig. 4 (A) The O 1s XPS spectra of the NiCo2O4 nanosheets. (B) Schematic illustration of the adsorption of H2O molecules onto the NiCo2O4 structure and the partial charge density of NiCo2O4 with oxygen vacancies. Reproduced with permission from ref. 28. Copyright 2015 Wiley-VCH. (C) O 1s XPS spectra of SC CoO NRs. (D) O–K edge XANES spectra of SC CoO NRs and reference CoO. (E) The projected density of states (PDOSs) on pristine CoO and CoO with O-vacancies. Reproduced with permission from ref. 32. Copyright 2016 Nature Publishing Group. | ||
Asnavandi et al. reported that the NiFe (oxy)hydroxides with surface oxygen vacancies achieve enhanced OER performance.31 X-ray absorption near edge structure (XANES) spectra displayed the relatively low oxidation states of Ni and Fe in NiFe–OOH, which may be associated with the presence of oxygen vacancies. Furthermore, to gauge the oxygen vacancy density, they fitted O 1s XPS spectra, which contains information of (1) lattice oxygen (530.1 eV), (2) oxygen loss in the material (531.7 eV), and (3) adsorbed oxygen throughout the surface (533.2 eV). The density of oxygen vacancy can be quantified roughly by the ratio of (2) to (1), and the density of NiFe with oxygen vacancies was 7.0, which is twice as high as that of the pristine one (3.2). NiFe with numerous vacancies exhibited enhanced OER current density (240 mA cm−2) at an overpotential of 270 mV from pristine NiFe (100 mA cm−2).
Ling et al. synthesized single-crystal cobalt oxide nanorods with exposed vacancy-rich pyramidal nano-facets.32 The O-vacancy formation was identified by the O 1s XPS spectra of single-crystal (SC) CoO nanorods (NRs) (Fig. 4C). The peak at the middle binding energy (II) at 531.5 eV was attributed to the oxygen vacant sites with relatively low oxygen coordination. The large area of peak II revealed that the SC CoO NRs contain numerous O-vacancies on the surface. Moreover, the peak at ∼536.0 eV of the O–K edge XANES spectra was associated with O deficiency, resulting in the presence of abundant O-vacancies on the surface of SC CoO NRs (Fig. 4D). This 1D SC CoO NRs exhibited an overpotential of 0.33 V (vs. RHE) in the OER. They explained that the conductivity of the metal oxides was further improved by creating O-vacancies on {111} facet and increasing the carrier concentration. Projected density of states (PDOS) on pristine CoO and CoO with O-vacancies was determined to examine the electronic structure, which correlated the formed O-vacancies in the bandgap of CoO to the observed strong adsorption of intermediates on the vacant sites and high electronic conductivity (Fig. 4E).
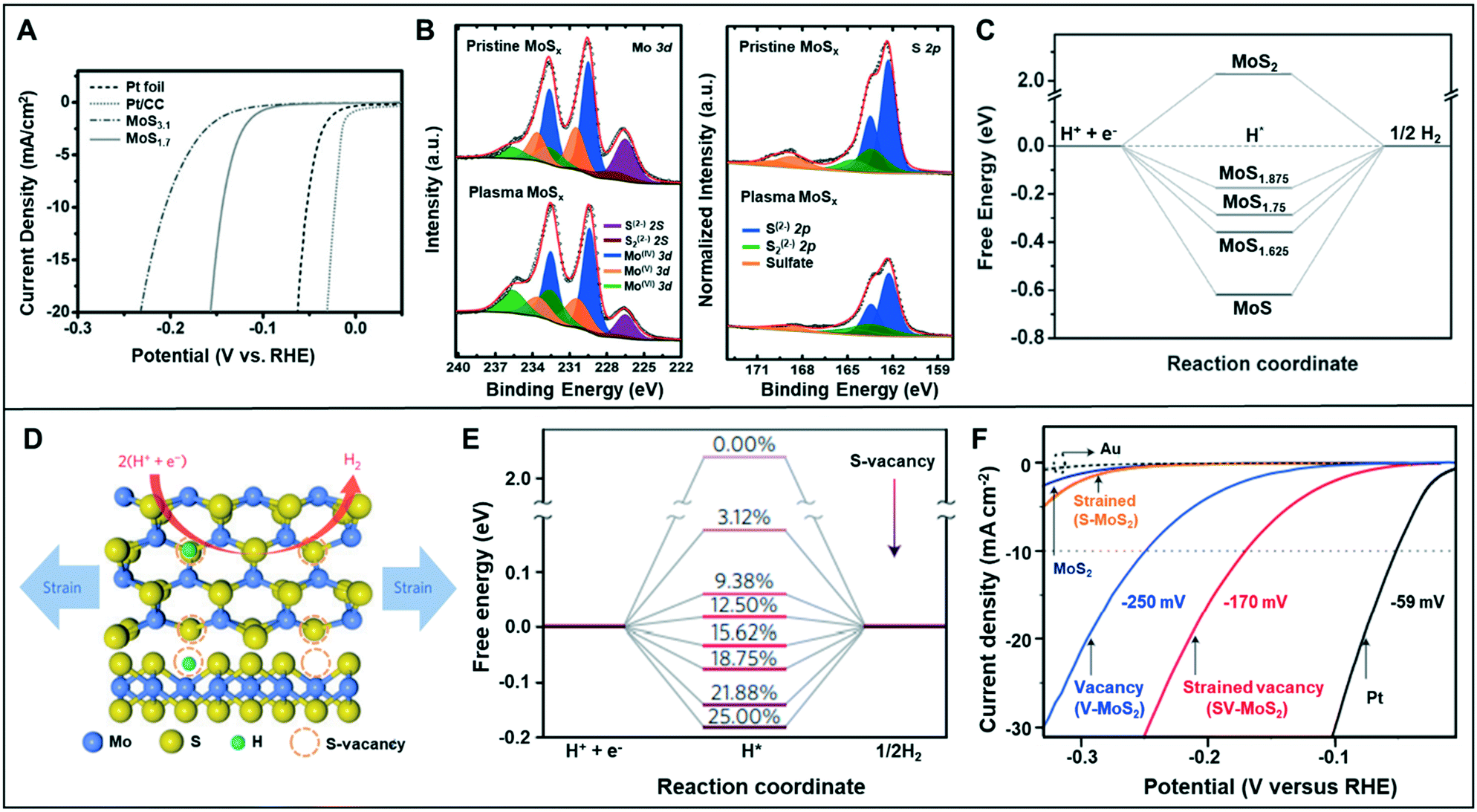 | ||
| Fig. 5 (A) HER polarization curves for Pt foil, Pt/CC, MoS3.1, and MoS1.7 in 0.5 M H2SO4 solution. (B) The Mo 3d and S 2p XPS spectra of the pristine MoSx and plasma MoSx. (C) The free energy with different defective levels of MoS2. Reproduced with permission from ref. 33. Copyright 2016 Wiley-VCH. (D) Schematic of the top (upper panel) and side (lower panel) views of MoS2 with strained S-vacancies on the basal plane. (E) Free energy versus the reaction coordinate of HER for the S-vacancy range of 0–25%. (F) LSV curves for Au substrate, Pt electrode, as-transferred MoS2 (strain: 0% and S-vacancy: 0%), strained MoS2 without S-vacancies (S-MoS2, strain: 1.35 ± 0.15% and S-vacancy: 0%), unstrained MoS2 with S-vacancies (V-MoS2, strain: 0% and S-vacancy: 12.5 ± 2.5%), and strained MoS2 with S-vacancies (SV-MoS2, strain: 1.35 ± 0.15% and S-vacancy: 12.5 ± 2.5%). Reproduced with permission from ref. 9. Copyright 2016 Nature Publishing Group. | ||
The placement of strain in the catalyst structure has been effective in optimizing the adsorption energy of the catalytic surface.34 Li et al. combined the sulfur vacancy with strain effect to optimize the basal plane of monolayer 2H–MoS2 (Fig. 5D).9 Catalysts with various amount of vacancies (0.00–25.00%) were analyzed by DFT calculations to understand the HER activity dependence on the vacancy (Fig. 5E). The calculation results verified that the free energy (ΔGH) related to hydrogen adsorption varied depending on the number of removed sulfur atoms. The free energy of pure 2H–MoS2 was about 2 eV, but it reduced to almost 0 eV at 12.50% of sulfur vacancy. Li et al. concluded that ΔGH might be reduced to 0 eV by applying the appropriate amount of strain in each case. The HER activity was measured with respect to the sulfur vacancy and strain-control in the catalyst (Fig. 5F). The modified catalyst with 12.5% S-vacancies and 1.35 ± 0.15% strain exhibited an overpotential of 170 mV at −10 mA cm−2 in HER, which was smaller than that of the catalyst modified with only S-vacancy (250 mV at −10 mA cm−2) or with only strain.
3.2. Cation vacancy
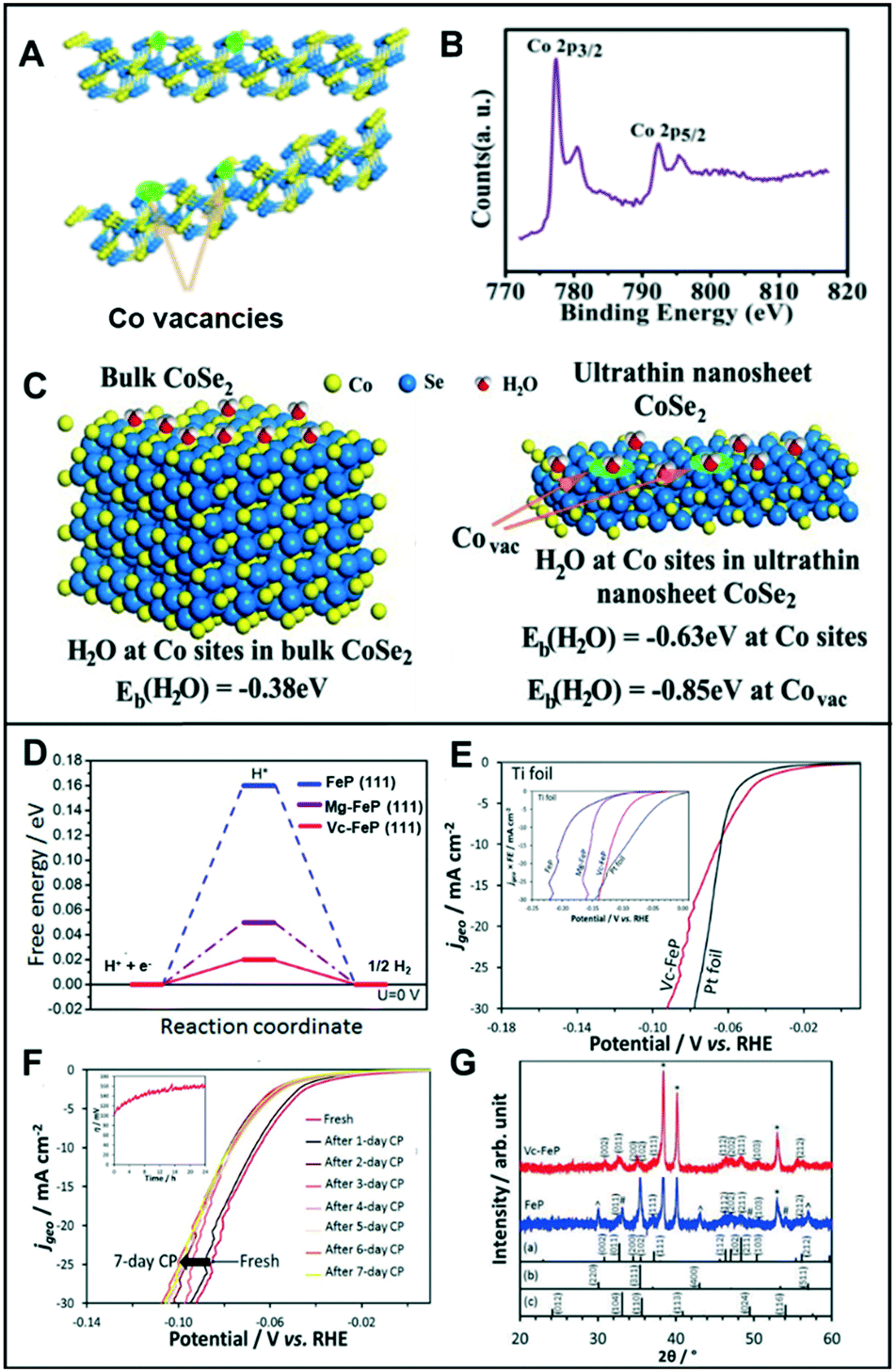 | ||
| Fig. 6 (A) Schematic illustration of the formation of Co vacancies in CoSe2 ultrathin nanosheets. (B) The XPS Co 2p of CoSe2 ultrathin nanosheets. (C) Geometries and binding energies of H2O molecules on Co sites and vacancies for bulk CoSe2 (left) and ultrathin nanosheet CoSe2 (right). Reproduced with permission from ref. 36. Copyright 2014 American Chemical Society. (D) Free energy diagram of FeP, Mg-FeP, and Vc-FeP (111) surfaces. (E) Polarization curves for the HER activity of Vc-FeP in 0.5 M H2SO4 (inset: HER activity in 1 M KOH). (F) The CP tests of Vc-FeP in 0.5 M H2SO4 (inset: stability test on Vc-FeP performed in 1 M KOH). (G) XRD data for FeP and Vc-FeP. The reference patterns of (a) orthorhombic FeP, (b) γ-Fe2O3, and (c) α-Fe2O3 also are shown. Reproduced with permission from ref. 37. Copyright 2017 Wiley-VCH. | ||
Likewise, Liu et al. utilized the appropriate hydrogen adsorption energy of the cation vacancies in five-fold twinned PtPdRuTe anisotropic structures (v-Pd3Pt29Ru62Te6 AS).39 The v-Pd3Pt29Ru62Te6 AS exhibited a low overpotential of 39 mV in 0.5 M H2SO4 and 22 mV in 1.0 M KOH to achieve current densities of −10 mA cm−2. They demonstrated that the activity of v-Pd3Pt29Ru62Te6 AS results from rapid charge transport and a substantial amount of exposed surface area of the anisotropic structure. The catalysts were stable for 30 h with a current density of −10 mA cm−2.
3.3. Multi-vacancies
A combination of the anion and cation vacancies often produces synergy for the enhanced electrochemical activity for water-splitting reaction.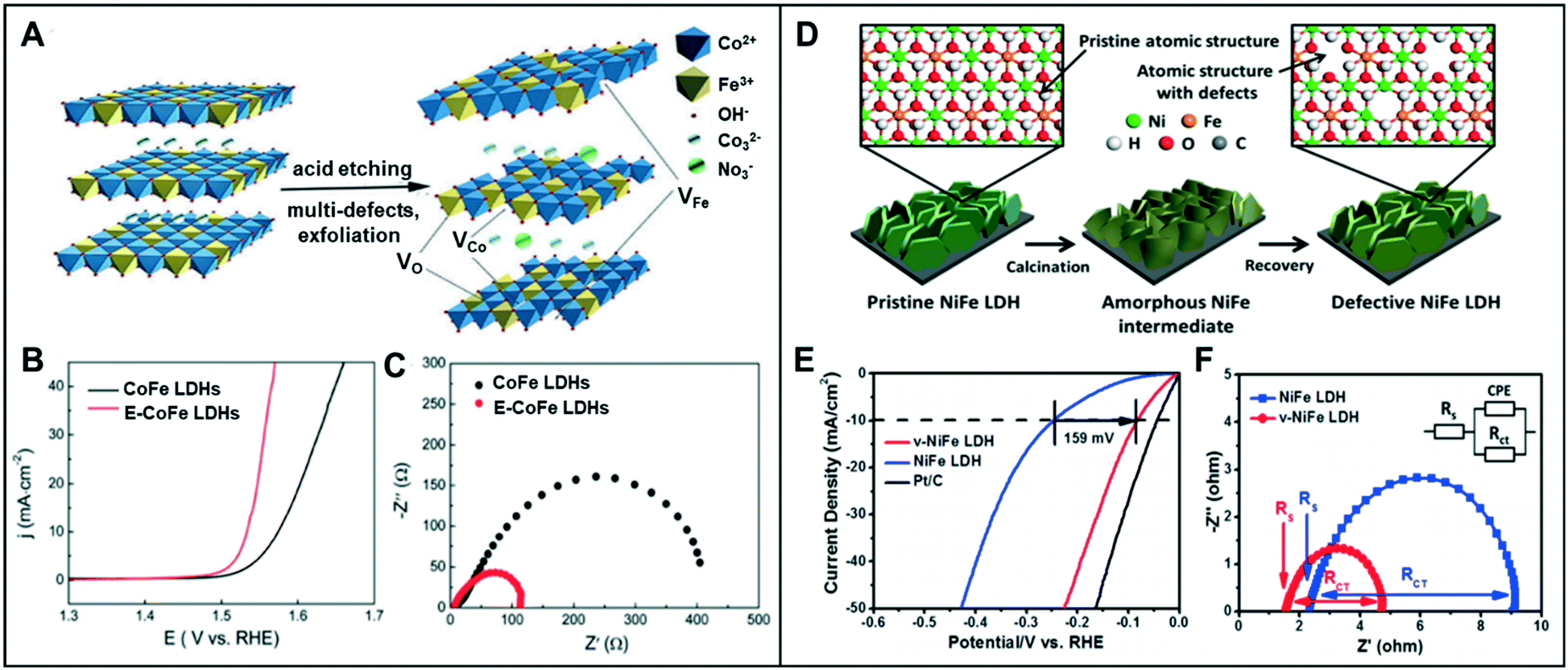 | ||
| Fig. 7 (A) Schematic illustration of E-CoFe LDHs after HNO3 etching. (B) OER performance of CoFe LDHs and E-CoFe LDHs in 1.0 M KOH. (C) Nyquist plots at an overpotential of 270 mV. Reproduced with permission from ref. 40. Copyright 2017 Royal Society of Chemistry. (D) Schematic illustration of defective NiFe LDH. (E) HER performance of v-NiFe LDH at a neutral pH. (F) Nyquist plots of pristine NiFe LDH and v-NiFe LDH nanosheets in 1.0 M PBS electrolyte. Reproduced with permission from ref. 42. Copyright 2019 American Chemical Society. | ||
Similarly, Wang et al. proved that the CoFe LDH nanosheets with multiple types of vacancies could promote the intrinsic activity in the OER.41 Furthermore, the ultrathin LDH nanosheets through Ar plasma etching (CoFe LDHs-Ar) resulted in the formation of multiple vacancies, including O, Co, and Fe vacancies. While the presence of oxygen vacancies facilitates the adsorption of intermediates such as OH*, O*, and OOH*, cationic vacancies cause the effective distributions of electrons and orbitals by adjusting the atomic environment of Co. With a potential increase in the valence state from Co2+ to Co3+, Co3+ with the higher valence state has a low coordination, resulting in increased catalytic activity. CoFe LDHs-Ar with multiple vacancies showed low overpotentials of 266 mV at 10 mA cm−2 and 313 mV at a relatively high current density of 50 mA cm−2 compared to that of CoFe LDHs (321 mV and 409 mV, respectively).
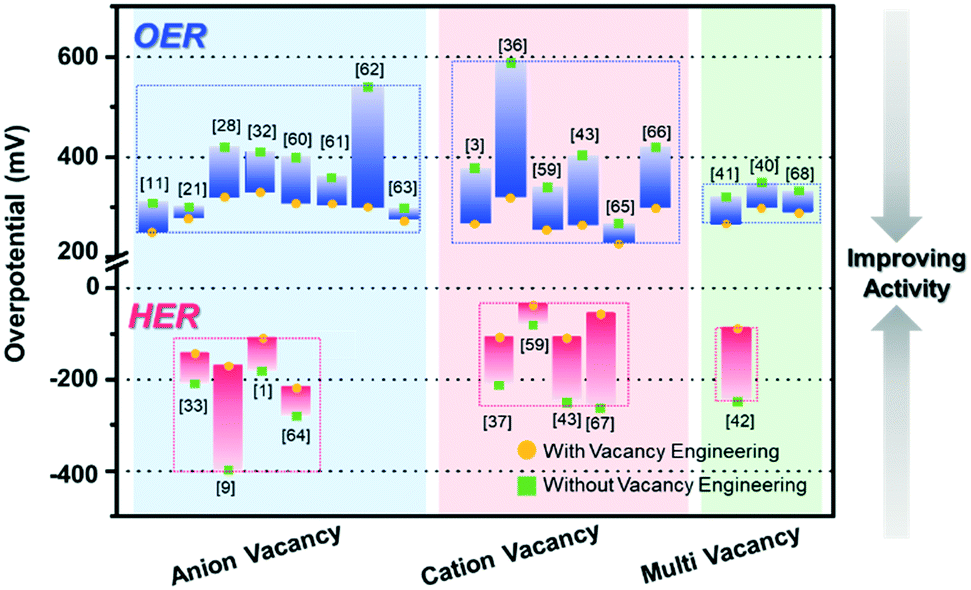
The electrocatalytic OER and HER performances of various catalysts and their vacancy types are described in Table 1. In addition, Fig. 8 summarizes the performance of recently reported vacancy-engineered electrocatalysts. The overpotentials are plotted against the types of vacancies to provide a visual illustration for the effective vacancy types toward the water-splitting reaction.
| Catalyst | Vacancy type | Application | Electrolyte | Overpotential [mV@10 mA cm−2] | Tafel slope [mV dec−1] | Ref. |
|---|---|---|---|---|---|---|
| SrCo0.9Ir0.1O3−δ | Anion vacancy | OER | 0.1 M HClO4 | 250 mV@1 mA cm−2 | — | 11 |
| IrNiOx | Anion vacancy | OER | 0.05 M H2SO4 | 280 mV@0.2 mA gIr−1 | — | 21 |
| NiCo2O4 nanosheets | Anion vacancy | OER | 1 M KOH | 320 mV | 30 | 28 |
| Reduced NiFe | Anion vacancy | OER | 1 M KOH | 270 mV | 40 | 31 |
| CoO nanorods | Anion vacancy | OER | 1 M KOH | 330 mV | 44 | 32 |
| Reduced Co3O4 NWs | Anion vacancy | OER | 1 M KOH | 420 mV@13.1 mA cm−2 | 72 | 26 |
| Fe1Co1-ONS | Anion vacancy | OER | 0.1 M KOH | 308 mV | 36.8 | 60 |
| CoOX(−4 h) nanoplates | Anion vacancy | OER | 1 M KOH | 306 mV | 67 | 61 |
| Plasma-engraved Co3O4 nanosheets | Anion vacancy | OER | 0.1 M KOH | 300 mV | 68 | 62 |
| YCRO-0.25 | Anion vacancy | OER | 0.5 M H2SO4 | 275 mV | 40.3 | 63 |
| Amorphous MoS1.7 | Anion vacancy | HER | 0.5 M H2SO4 | 143 mV | 39.5 | 33 |
| MoS2 basal plane | Anion vacancy | HER | 0.5 M H2SO4 | 170 mV | 60 | 9 |
| NiO NRs-m-Ov | Anion vacancy | HER | 1 M KOH | 110 mV | 100 | 1 |
| Mesoporous MoO3−x | Anion vacancy | HER | 0.1 M KOH | 138 mV | 56 | 15 |
| MoS2−xNy/rGO | Anion vacancy | HER | 0.5 M H2SO4 | 217 mV | 48 | 64 |
| NiFe-VM-O | Cation vacancy | OER | 1 M NaOH | 371 mV | 28 | 22 |
| Co3−xO4 | Cation vacancy | OER | 1 M KOH | 268 mV | 38.2 | 3 |
| CoSe2 nanosheets | Cation vacancy | OER | 0.1 M KOH | 320 mV | 44 | 36 |
| NiFe LDHs-VNi nanosheets | Cation vacancy | OER | 1 M KOH | 229 mV | 62.9 | 65 |
| SnCoFe-Ar | Cation vacancy | OER | 1 M KOH | 300 mV | 42.3 | 66 |
| FeP | Cation vacancy | HER | 1 M KOH | 108 mV | 62 | 37 |
| FeP | Cation vacancy | HER | 0.5 M H2SO4 | 65 mV | 49 | 37 |
| PtPdRuTe AS | Cation vacancy | HER | 1 M KOH | 22 mV | 22 | 39 |
| Pt/np-Co0.85Se | Cation vacancy | HER | 1 M PBS | 55 mV | 35 | 67 |
| NiAlδP | Cation vacancy | HER | 0.5 M H2SO4 | 35 mV | 38 | 59 |
| OER | 0.5 M H2SO4 | 256 mV | 76 | 59 | ||
| δ-FeOOH NSs/NF | Cation vacancy | HER | 1 M KOH | 108 mV | 68 | 43 |
| OER | 1 M KOH | 265 mV | 36 | 43 | ||
| CoFe LDHs-Ar nanosheets | Multi vacancy | OER | 1 M KOH | 266 mV | 37.85 | 41 |
| E-CoFe LDHs | Multi vacancy | OER | 1 M KOH | 300 mV | 41 | 40 |
| NiFe-LDH nanosheets | Multi vacancy | OER | 1 M KOH | 254 mV | 32 | 27 |
| H2O-Plasma exfoliated CoFe LDH | Multi vacancy | OER | 1 M KOH | 290 mV | 36 | 68 |
| NiFe LDHs | Multi vacancy | HER | 1 M PBS | 87 mV | 46.3 | 42 |
 | ||
| Fig. 8 Trend of the performance of recent vacancy-engineered electrocatalysts. | ||
In general, the vacancies are classified into three types, namely anion vacancies, cation vacancies, and multi-vacancies. Anion vacancies are more easily formed, and oxygen vacancies are commonly observed with formed oxide-based electrocatalysts for both the OER and HER. In contrast, the formation of the cation vacancy is considerably challenging due to the high formation energy required, compared to the case with the anion vacancy.39 As compared to the electrocatalysts without vacancies, the cation vacancy-engineered electrocatalysts remarkably lowered the overpotentials, and thus showed improved activity toward the OER and HER, as shown in Fig. 8, owing to their diverse chemical states that are conducive for the optimization of intermediate B.Es For the HER, the cation vacancy-controlled catalysts performed much better than the anion vacancy-engineered catalysts, with ∼50 mV lower overpotentials.
Multi-vacancies with both cation and anion vacancies showed the synergistic effects of cation and anion vacancies toward enhancing the performances of the electrocatalysts. The presence of anion vacancies can effectively modulate the electronic structures, tune the adsorption energies of intermediates, and improve the electrical conductivity.41 The presence of cation/metal vacancies can increase the valence state of nearby metal centers, which can also improve the reaction kinetics due to the increased surface area with a low CN of the catalytic active sites.42 However, the examples for multi-vacancies are very limited, thus calling for a great synthetic effort to the controlled formation of multi-vacancies in catalytic materials.
For practical applications of the electrocatalysts, their stability, the intrinsic property dependent on the material phases, should be attained.43 Vacancy engineering of electrocatalysts might provide solutions to the catalyst stability issues via optimized adsorption energy of intermediates and improved charge transfer in the catalyst structure. Some vacancy engineered catalysts with the high concentration of oxygen vacant sites have shown a superior stability toward OER, because the oxygen-vacant sites can form direct O–O bonds in the OER process, not necessitating significant reorganization of atoms within the catalysts.29 Vacancy of other anions such as sulfur anions can also boost the catalyst stability; porous nanosheets of Co3S4 with abundant sulfur vacancies showed excellent stability toward the HER even at the high negative potentials.44 Catalysts with sulfur vacancies, having more electrons, showed lower adsorption energy of H2O, compared to other catalysts with no vacancy, accelerating electron/mass transfer and thus enhancing the stability.
However, excessive vacancies in the catalysts could also induce adverse effects on structural stability and electronic conductivity.45,46 Also, the issues for structural degradation and dissolution through vacant sites are far from being resolved. Therefore, the establishment of relationships between the catalytic stability and the number and nature of vacancies of the catalysts remains an important task in accomplishing the catalyst stability toward water splitting.
4. Future perspectives
The rapid growth of interest in engineering vacancy-rich electrocatalysts is a response to the increasing demand for highly efficient electrocatalysts. In this review, we aimed at providing an overview of the recent studies on vacancy-engineered catalysts for electrochemical HER and OER, regardless of metal types. These engineering vacancies in electrocatalysts could lead to the exposure of active sites through CUSs, optimization of adsorption energy for intermediates, and improvement of electrical conductivity. Based on these features from vacancy-engineered electrocatalysts, we could categorize the types of vacancies into anion, cation, and multi-vacancies. Despite the significant achievements in vacancy engineering toward water splitting in this review, the synthetic methodologies for the controlled formation of different vacancy types are severely underdeveloped. Therefore, continuous efforts on understanding the relationship between vacancy formation and catalytic performance are necessary to rationally design vacancy-optimized electrocatalysts. Some representative future perspectives are schematically shown in Fig. 9.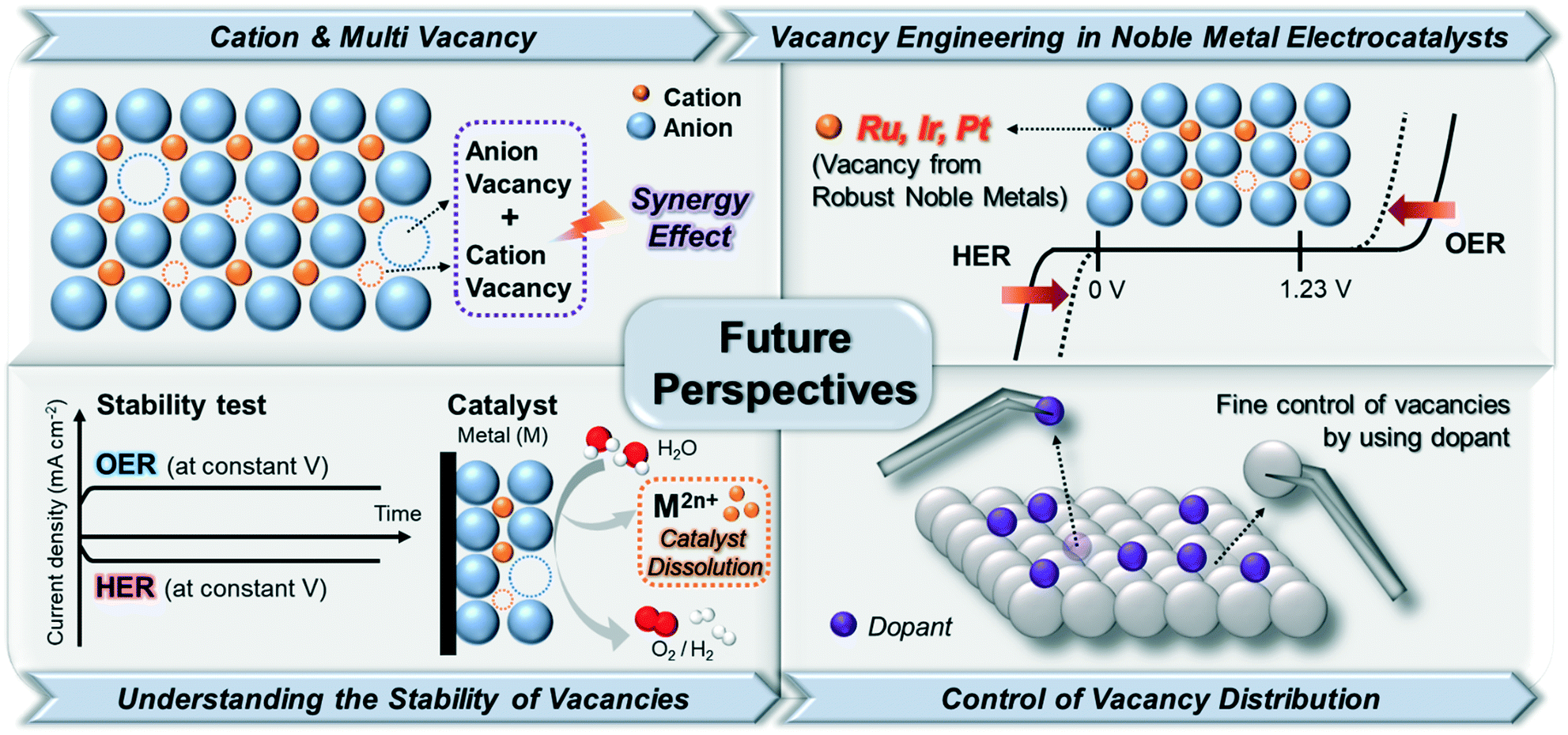 | ||
| Fig. 9 Illustration of future perspectives for enhancing the performance of electrocatalytic water splitting via vacancy engineering. | ||
4.1. Beyond anion vacancy engineering: importance of cation and multi-vacancy
The most remarkable challenges for vacancy engineering are the limited methods to induce cation vacancies because the formation energy of metal/cation vacancy is quite higher than that of anion vacancy.47 However, as discussed in Table 1 and Fig. 8, the cation or metal vacancy-engineered catalysts could largely improve the catalytic activity for both OER and HER. This is because the excess electrons can facilitate the modulation of the electronic structure on the catalyst surface. Therefore, additional research to create metal/cation vacancies to generate OER and HER active sites on the atomic level should be conducted. Moreover, several studies have proven that multi (anion and cation) vacancies within one material can have synergistic effects to accelerate the water electrolysis process. In this context, the engineering of multi-vacancies with various catalytic materials is direly required for enhancing electrochemical water-splitting reactions.4.2. Development of a novel vacancy from noble metal-based electrocatalysts
The development of noble metal vacancies is also necessary. In acidic OER/HER, noble metals such as Ir, Ru, and Pt have been greatly utilized because of their excellent intrinsic performances for electrolysis. Although the dissolution rates of noble metals during electrochemical catalysis are lower than those of non-noble metals, a small amount of noble metals can still be traced in the electrolyzers.11,48 Although such dissolution can initiate defects in the electrocatalysts with noble metal vacancies, these vacancies could not be finely controlled. The formation of well-defined noble metal vacancies can lead to the great modification of metal coordination, which can provide additional active sites and improve the electrocatalytic performance of water splitting in both acidic and alkaline media using a relatively small amount of noble metals. Thus, further studies on noble metal-based vacancies are required.4.3. Constructing the stability of vacancy-engineered electrocatalysts
Very few studies have been conducted to understand the relationship between the formation of vacancies and catalyst stability. Because the vacancies in the structures can lead to geometrical and electronic distortions,49 a weaker bonding of the lattice in defective structures compared to that in crystalline ones might be induced, which would lead to the formation of metastable structures and accelerate the degradation of catalyst structures in the electrocatalysts.48 To prevent the degradation of electrocatalysts, an understanding of the surrounding atoms adjacent to the vacant sites and their overall chemical environments is necessary to design highly robust electrocatalysts with numerous active sites. Although a few studies on the theoretical investigation of thermodynamic stability and vacancy formation has been reported,50–52 experimental research demonstrating the relationship between an optimal vacancy formation and stability of electrocatalysts should also be conducted to verify the theory-based predictions.4.4. Efforts on the elaborate control of the vacancy distribution
To aid the development of vacancy formation, advances in synthetic methods (both physical and chemical) to form and identify vacancies are greatly required. The energy to create vacancies can be provided by physical methods, such as plasma etching,9 thermal treatment,53 liquid exfoliation,27,54 and electrochemical etching.11,21 These physical methods used to create vacancies could be modulated by varying the processing time and energy input.18 Meanwhile, the creation of vacancies can occur simultaneously with chemical reactions, such as chemical reduction,55 hydrothermal synthesis,56 and elemental doping.57 Therefore, because it is difficult to predict and precisely control the vacancy distribution, elemental doping into existing materials might be an effective route to create atomically controlled vacancies.58 Depending on the type of dopants with different ionic sizes, the positions and sizes of the formed vacancy sites could be deterministically controlled. For example, in perovskites (ABO3, where A and B are cations, with A atoms larger than B atoms; X is an anion that bonds with both cations), small ions are exclusively doped on the B-site and oxygen vacancies are created for charge composition if the dopant has a relatively low valence.57,58 This can also result in the partial oxidation or reduction of ions on the B sites or result in the appearance of B-site vacancies if the dopant has a high valence. Conversely, if large ions are substituted only on the A-site, A-site, B-site, or oxygen vacancies can be created, depending on the induced charge balance of the crystal structure. Therefore, the choice of doping element types can determine the different sizes and sites of vacancies, which can lead to the discovery of more active sites on the electrocatalysts. As with the case of perovskites, the atomically modulated vacancies via elemental doping might be applied to various types of crystal structures, such as hetero-interface structures. Nanostructures with hetero-interfaces can be promising platforms for providing a pathway through which dopants can easily move between different phases under relatively low energy, compared to nanostructures without interfaces. This might lead to the formation of numerous vacancies after leaching out the dopants. The position and number of vacancies might be conveniently controlled by modulating the diffusion zone width within the nanocrystals.58Furthermore, even if the materials have the same crystal structure and geometry, the shape-controlled materials might have different properties when the vacancy is formed on the exposed surface. Shape-controlled materials that expose the different specific facets can have facet-specific vacancy types on the surface of the catalysts due to the differences in the coordination environments for different facet types. Investigation of catalysis on facet-specific vacancy might allow us to understand further structural details of the highly active vacancies toward water electrolysis.
5. Conclusion
In summary, vacancy engineering has been considered as an effective strategy for the development of highly efficient electrocatalysts for water electrolysis. The vacancy-controlled catalysts are endowed with useful properties such as exposure of low-coordination sites, optimized adsorption energies of intermediates, and enhanced electrical conductivity. In addition, the vacancy-engineered electrocatalysts show great potential toward the water-splitting reactions, namely OER and HER. However, synthetic approaches are quite underdeveloped toward position-controlled and quantity-controlled vacancy formation within the nanostructures. Therefore, the development of atomic-scale engineering for the precise tuning of vacancies and alteration of coordination environments for vacancies should be developed for further improvement of active and stable electrocatalysts. We hope that this review can motivate active developments of vacancy-engineered electrocatalysts for water electrolysis and other related energy applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF-2017R1A2B3005682, NRF2019R1A6A1A11044070), and the Hydrogen Energy Innovation Technology Development Program of the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science and ICT(MSIT)) (No. NRF-2019M3E6A1064709).References
- T. Zhang, M.-Y. Wu, D.-Y. Yan, J. Mao, H. Liu, W.-B. Hu, X.-W. Du, T. Ling and S.-Z. Qiao, Nano Energy, 2018, 43, 103–109 CrossRef CAS.
- Z. Chen, C. X. Kronawitter, Y.-W. Yeh, X. Yang, P. Zhao, N. Yao and B. E. Koel, J. Mater. Chem. A, 2017, 5, 842–850 RSC.
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J.-J. Zou, ACS Catal., 2018, 8, 3803–3811 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- S. H. Brodersen, U. Grønbjerg, B. Hvolbæk and J. Schiøtz, J. Catal., 2011, 284, 34–41 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- J. V. Lauritsen, J. Kibsgaard, S. Helveg, H. Topsøe, B. S. Clausen, E. Lægsgaard and F. Besenbacher, Nat. Nanotechnol., 2007, 2, 53–58 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- S. Jayabal, G. Saranya, J. Wu, Y. Liu, D. Geng and X. Meng, J. Mater. Chem. A, 2017, 5, 24540–24563 RSC.
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef PubMed.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- Y. Liu, C. Xiao, P. Huang, M. Cheng and Y. Xie, Chem, 2018, 4, 1263–1283 CAS.
- Z. Luo, R. Miao, T. D. Huan, I. M. Mosa, A. S. Poyraz, W. Zhong, J. E. Cloud, D. A. Kriz, S. Thanneeru, J. He, Y. Zhang, R. Ramprasad and S. L. Suib, Adv. Energy Mater., 2016, 6, 1600528 CrossRef.
- Y.-W. Cheng, J.-H. Dai, Y.-M. Zhang and Y. Song, J. Mater. Chem. A, 2018, 6, 20956–20965 RSC.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- M.-Q. Yang, J. Wang, H. Wu and G. W. Ho, Small, 2018, 14, 1703323 CrossRef PubMed.
- K. S. Exner and H. Over, ACS Catal., 2019, 9, 6755–6765 CrossRef CAS.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Velasco Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7, 6791–6795 RSC.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- H. J. Lee, S. Back, J. H. Lee, S. H. Choi, Y. Jung and J. W. Choi, ACS Catal., 2019, 9, 7099–7108 CrossRef CAS.
- H.-S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS PubMed.
- M. Dieterle, G. Weinberg and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 812–821 RSC.
- G. Wang, Y. Ling and Y. Li, Nanoscale, 2012, 4, 6682–6691 RSC.
- Y. Wang, T. Zhou, K. Jiang, P. Da, Z. Peng, J. Tang, B. Kong, W.-B. Cai, Z. Yang and G. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- Y. Zhao, X. Zhang, X. Jia, G. I. N. Waterhouse, R. Shi, X. Zhang, F. Zhan, Y. Tao, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2018, 8, 1703585 CrossRef.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- S. Hirai, K. Morita, K. Yasuoka, T. Shibuya, Y. Tojo, Y. Kamihara, A. Miura, H. Suzuki, T. Ohno, T. Matsuda and S. Yagi, J. Mater. Chem. A, 2018, 6, 15102–15109 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- M. Asnavandi, Y. Yin, Y. Li, C. Sun and C. Zhao, ACS Energy Lett., 2018, 3, 1515–1520 CrossRef CAS.
- T. Ling, D.-Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X.-W. Du, Z. Hu, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2016, 7, 12876 CrossRef CAS PubMed.
- A.-Y. Lu, X. Yang, C.-C. Tseng, S. Min, S.-H. Lin, C.-L. Hsu, H. Li, H. Idriss, J.-L. Kuo, K.-W. Huang and L.-J. Li, Small, 2016, 12, 5530–5537 CrossRef CAS PubMed.
- B. T. Sneed, A. P. Young and C.-K. Tsung, Nanoscale, 2015, 7, 12248–12265 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- W. L. Kwong, E. Gracia-Espino, C. C. Lee, R. Sandström, T. Wågberg and J. Messinger, ChemSusChem, 2017, 10, 4544–4551 CrossRef CAS PubMed.
- W. L. Kwong, C. C. Lee and J. Messinger, J. Phys. Chem. C, 2017, 121, 284–292 CrossRef CAS.
- S. Liu, X. Mu, W. Li, M. Lv, B. Chen, C. Chen and S. Mu, Nano Energy, 2019, 61, 346–351 CrossRef CAS.
- P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2017, 53, 11778–11781 RSC.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- Z. Yuan, S.-M. Bak, P. Li, Y. Jia, L. Zheng, Y. Zhou, L. Bai, E. Hu, X.-Q. Yang, Z. Cai, Y. Sun and X. Sun, ACS Energy Lett., 2019, 4, 1412–1418 CrossRef CAS.
- J. Kim, M. Jun, S. Choi, J. Jo and K. Lee, Nanoscale, 2019, 11, 20392–20410 RSC.
- C. Zhang, Y. Shi, Y. Yu, Y. Du and B. Zhang, ACS Catal., 2018, 8, 8077–8083 CrossRef CAS.
- J. Du, T. Zhang, F. Cheng, W. Chu, Z. Wu and J. Chen, Inorg. Chem., 2014, 53, 9106–9114 CrossRef CAS PubMed.
- Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS PubMed.
- B. Liu, Y. Wang, H.-Q. Peng, R. Yang, Z. Jiang, X. Zhou, C.-S. Lee, H. Zhao and W. Zhang, Adv. Mater., 2018, 30, 1803144 CrossRef PubMed.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- A. A. Emery and C. Wolverton, Sci. Data, 2017, 4, 170153 CrossRef CAS PubMed.
- B. Liu, D. S. Aidhy, Y. Zhang and W. J. Weber, Phys. Chem. Chem. Phys., 2014, 16, 25461–25467 RSC.
- S. M. Alay-e-Abbas, S. Nazir, N. A. Noor, N. Amin and A. Shaukat, J. Phys. Chem. C, 2014, 118, 19625–19634 CrossRef CAS.
- D. Le, T. B. Rawal and T. S. Rahman, J. Phys. Chem. C, 2014, 118, 5346–5351 CrossRef CAS.
- L. Li, T. Zhang, J. Yan, X. Cai and S. Liu, Small, 2017, 13, 1700441 CrossRef PubMed.
- Y. Xu, L. Wang, X. Liu, S. Zhang, C. Liu, D. Yan, Y. Zeng, Y. Pei, Y. Liu and S. Luo, J. Mater. Chem. A, 2016, 4, 16524–16530 RSC.
- C. Zhu, S. Fu, D. Du and Y. Lin, Chem. – Eur. J., 2016, 22, 4000–4007 CrossRef CAS PubMed.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef.
- Y. Wang, J. Piao, G. Xing, Y. Lu, Z. Ao, N. Bao, J. Ding, S. Li and J. Yi, J. Mater. Chem. C, 2015, 3, 11953–11958 RSC.
- G. Li, G. R. Blake and T. T. M. Palstra, Chem. Soc. Rev., 2017, 46, 1693–1706 RSC.
- W. Cheng, H. Zhang, X. Zhao, H. Su, F. Tang, J. Tian and Q. Liu, J. Mater. Chem. A, 2018, 6, 9420–9427 RSC.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- W. Xu, F. Lyu, Y. Bai, A. Gao, J. Feng, Z. Cai and Y. Yin, Nano Energy, 2018, 43, 110–116 CrossRef CAS.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- Q. Feng, Z. Zhao, X.-Z. Yuan, H. Li and H. Wang, Appl. Catal., B, 2020, 260, 118176 CrossRef CAS.
- Z. Luo, Y. Wang, X. Wang, Z. Jin, Z. Wu, J. Ge, C. Liu and W. Xing, ACS Appl. Mater. Interfaces, 2019, 11, 39782–39788 CrossRef CAS PubMed.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 14, 1800136 CrossRef PubMed.
- D. Chen, M. Qiao, Y.-R. Lu, L. Hao, D. Liu, C.-L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691–8696 CrossRef CAS PubMed.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |