
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermodynamically stable and metastable coordination polymers synthesized from solution and the solid state†
Tristan
Neumann
a,
Gianpiero
Gallo
*bc,
Inke
Jess
a,
Robert E.
Dinnebier
 b and
Christian
Näther
*a
b and
Christian
Näther
*a
aInstitute of Inorganic Chemistry, Kiel University, Max-Eyth-Straße 2, 24118 Kiel, Germany. E-mail: cnaether@ac.uni-kiel.de
bMax-Planck-Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany. E-mail: G.Gallo@fkf.mpg.de
cDepartment of Chemistry and Biology “A. Zambelli”, University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy
First published on 4th November 2019
Abstract
The reaction of Fe(NCS)2 with 4-picoline in solution leads to two new discrete complexes, Fe(NCS)2(4-picoline)4·solvate (1-Fe) and Fe(NCS)2(4-picoline)2(H2O)2 (2-Fe–H2O). Upon heating, both compounds transform into two isomers with the composition [Fe(NCS)2(4-picoline)2]n (2-Fe/I and 2-Fe/II) that consist of chains and that are related by monotropy, with 2-Fe/I as the thermodynamically stable form. Upon further heating, a transformation into a more condensed chain compound of unusual structure with the composition [Fe(NCS)2(4-picoline)]n (3-Fe) is observed. The crystal structures of 2-Fe/I and 2-Fe/II were determined by Rietveld refinements, as it was found that 2-Fe/I is isotypic to the corresponding Cd compound 2-Cd/I, whereas 2-Fe/II is isotypic to [Cu(NCS)2(4-cyanopyridine)2]n which was reported recently. To retrieve structural information on 3-Fe the corresponding Cd compound (3-Cd) was synthesized and was shown to be isotypic to 3-Fe. Finally, upon heating the already known compounds [Cd(NCS)2(4-picoline)4]·0.67·4-picoline·0.33·H2O solvate (1-Cd) and [Cd(NCS)2(4-picoline)2]n (2-Cd/I) a new thermodynamically metastable modification was found (2-Cd/III) that is isotypic to [Cd(NCS)2(4-cyanopyridine)2]n and behaves monotropically to 2-Cd/I with the latter as the thermodynamically stable phase.
Introduction
The synthesis of new coordination polymers, inorganic–organic hybrid compounds or metal organic frameworks is still an important goal in modern coordination chemistry because several of them have a lot of potential for applications.1–6 Common to all of these materials is that they are typically synthesized by reactions in solution at room or elevated temperatures. However, there are alternative routes as well, like synthesis through molecular milling, molten flux synthesis, solid–gas reactions or by thermal decomposition of suitable precursors.7–21 The usage of these methods can have numerous advantages because no solvent is needed; very often they proceed in quantitative yield and lead in an irreversible reaction to the formation of pure samples. Moreover, in several cases crystalline phases are accessible that cannot be prepared in solution because they are metastable, including polymorphic or isomeric modifications.22–28 This is the case, e.g., for thermal decomposition reactions of precursor compounds based on transition metal thio- and selenocyanates. If they consist of less chalcophilic metal cations, most of these compounds contain only terminally N-bonded anionic ligands, since the additional coordination to sulfur, leading to the formation of a bridging coordination, is unfavourable.29–35 This is a pity, especially because the bridging coordination is of interest, as it will lead to coordination polymers of different dimensionality, which is of special importance, e.g., for the synthesis of magnetic materials.36–43 This is completely different for coordination polymers based on chalcophilic cations like Cd(II), which easily forms compounds with bridging thiocyanate anions.44,45 However, if such compounds with terminally anionic ligands are heated, the coligands are often emitted in discrete steps, which enforces the formation of intermediates with condensed thiocyanate networks.10,46–48 In several cases, polymorphic or isomeric modifications are obtained, which is of importance for the study of structure–property relationships.49–51 By this reaction, crystalline powders not suitable for single crystal X-ray diffraction (SCXRD) are obtained, but in this case, the corresponding Cd compounds can easily be crystallized from solution (see above) and in several cases they are isotypic to the less chalcophilic metal cations, which allows the retrieval of structural information by Rietveld refinements.52Recently, we became especially interested in Fe(II) thiocyanate compounds, which, in contrast to, e.g. Mn(II), Co(II) and Ni(II), are much more difficult to prepare. In several cases only simple discrete complexes like aqua complexes are obtained and Fe(II) compounds with a bridging coordination or more condensed thiocyanate networks are very rare. This is the case for, e.g. Fe(NCS)2 coordination compounds with 4-picoline as a coligand, for which only a discrete anionic complex with the composition [4-picolineH]22+ [Fe(NCS)2(4-picoline)4]2− and one solvate with the composition [Fe(NCS)2(4-picoline)4]·4-picoline are reported.53,54 We additionally obtained two discrete complexes from solution containing one solvate and an aqua complex. However, measurements using thermogravimetry and differential scanning calorimetry (TG-DSC) and temperature dependent XRPD measurements indicate the formation of two new isomeric modifications, consisting of chains, which were investigated for their thermodynamic relations. Surprisingly, upon further heating, a more condensed compound is obtained showing a rare chain topology, which for Fe(II) was never reported before. Here we report on these investigations.
Results
Synthetic aspects and thermoanalytical investigations
The reaction of FeCl2·4H2O with KSCN and 4-picoline in different solvents like, e.g., water, methanol or ethanol only leads to the formation of two new crystalline phases (1-Fe and 2-Fe/H2O) and the discrete complex [Fe(NCS)2(4-picoline)4]·4-picoline already reported in the literature (see Experimental).53 For 1-Fe the CN stretch is found much below 2100 cm−1 in the IR and the Raman spectra, indicating the presence of only terminally anionic ligands (Fig. S1†).55,56 For 2-Fe/H2O, slightly higher values are observed that are at the borderline between a terminal and a bridging coordination (Fig. S2†). Elemental analysis of 1-Fe suggests a ratio between Fe(NCS)2 and 4-picoline of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 but later it was found that this compound contains a small amount of solvent (see Experimental). 2-Fe/H2O forms an aqua complex with the composition Fe(NCS)2(4-picoline)2(H2O)2. Crystals of 1-Fe were obtained and characterized by SCXRD. This compound crystallizes in the trigonal (rhombohedral setting) space group R
:4 but later it was found that this compound contains a small amount of solvent (see Experimental). 2-Fe/H2O forms an aqua complex with the composition Fe(NCS)2(4-picoline)2(H2O)2. Crystals of 1-Fe were obtained and characterized by SCXRD. This compound crystallizes in the trigonal (rhombohedral setting) space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with Z = 9 formula units in the unit cell. The Fe(II) cations are octahedrally coordinated by four 4-picoline coligands and two terminal N-bonded thiocyanate anions (Fig. S3†). The complexes are arranged in a way that channels containing disordered solvent molecules are formed along the crystallographic c-axis (Fig. S4†). For 2-Fe/H2O no single crystals were obtained and therefore its structure was determined ab initio by X-ray powder diffraction (XRPD; Fig. S5†). This compound crystallizes in the monoclinic space group P21/c with Z = 2 formula units in the unit cell. The Fe(II) cations are octahedrally coordinated by two terminal N-bonded thiocyanate anions, two 4-picoline coligands and two water molecules (Fig. S6†). XRPD measurements prove that 1-Fe, [Fe(NCS)2(4-picoline)4]·4-picoline and 2-Fe/H2O were obtained as pure phases (Fig. S7–S9†).
with Z = 9 formula units in the unit cell. The Fe(II) cations are octahedrally coordinated by four 4-picoline coligands and two terminal N-bonded thiocyanate anions (Fig. S3†). The complexes are arranged in a way that channels containing disordered solvent molecules are formed along the crystallographic c-axis (Fig. S4†). For 2-Fe/H2O no single crystals were obtained and therefore its structure was determined ab initio by X-ray powder diffraction (XRPD; Fig. S5†). This compound crystallizes in the monoclinic space group P21/c with Z = 2 formula units in the unit cell. The Fe(II) cations are octahedrally coordinated by two terminal N-bonded thiocyanate anions, two 4-picoline coligands and two water molecules (Fig. S6†). XRPD measurements prove that 1-Fe, [Fe(NCS)2(4-picoline)4]·4-picoline and 2-Fe/H2O were obtained as pure phases (Fig. S7–S9†).
The two new precursor compounds 1-Fe and 2-Fe/H2O were investigated by TG-DSC measurements to check if 4-picoline deficient phases might be accessible. Upon heating, three mass loss steps are observed in the TG curve, which are accompanied by endothermic events in the DSC curve (Fig. 1, S10 and S11†). The experimental mass losses in the first steps are in reasonable agreement with those calculated for the removal of two 4-picoline ligands for 1-Fe (Δmcalc = −34.2%) and two water molecules for 2-Fe/H2O (Δmcalc = −9.1%), indicating that compounds with the composition [Fe(NCS)2(4-picoline)2]n (2-Fe) have formed. The mass losses in the second TG step point to the removal of a further 4-picoline ligand (Δmcalc = −17.1% without solvate molecules for 1-Fe and Δmcalc = −23.6% for 2-Fe/H2O), leading to compounds with the composition [Fe(NCS)2(4-picoline)]n (3-Fe), and in the last step the remaining coligands are emitted, leading to the formation of Fe(NCS)2. These results indicate that 1-Fe and 2-Fe/H2O transform into new compounds with the composition [Fe(NCS)2(4-picoline)2]n (2-Fe/I) as intermediates, which, on further heating, form phases with the composition [Fe(NCS)2(4-picoline)]n (3-Fe). The second TG step is poorly resolved. Therefore, heating rate dependent measurements were performed, showing no improvement in the resolution (Fig. S10 and S11†).
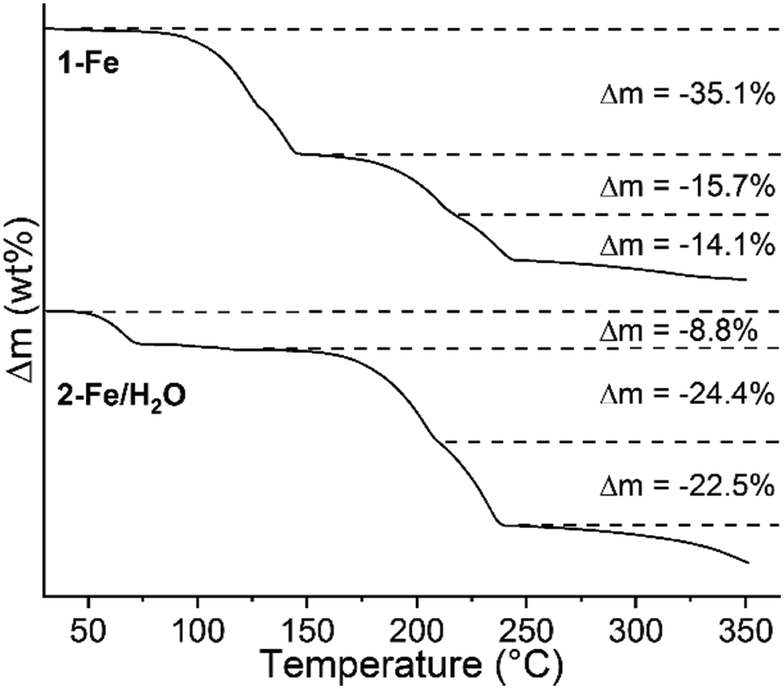 | ||
| Fig. 1 TG curves of 1-Fe (top) and 2-Fe/H2O (bottom) measured at a heating rate of 1 °C min−1, together with the relative experimental mass loss. | ||
To identify the new intermediate compounds, the residues obtained after the respective mass loss steps were investigated by XRPD. This shows that always good crystalline samples have formed and that the phases obtained after each mass loss of 1-Fe and 2-Fe/H2O are identical (Fig. S12†). Unfortunately, 2-Fe/I cannot be prepared from solution, and therefore no single crystals were available. However, based on its composition and IR/Raman analyses (the CN stretch is observed at 2090 cm−1 in the IR and at 2092 cm−1 in the Raman spectra; Fig. S13†), it is highly likely that it will consist of chains, in which the cations are linked by pairs of anionic ligands, as this is a very common motif in this class of compounds. Comparison of XRPD patterns of 2-Fe/I with the thiocyanate coordination polymers having 4-picoline as coligand revealed similarities to the chain compound [Cd(NCS)2(4-picoline)2]n already reported in the literature (Fig. S14†)57,58 Therefore, a Rietveld refinement was performed, which proves that [Fe(NCS)2(4-picoline)2]n (2-Fe/I) is isotypic to its Cd analog (Fig. S15†).
To obtain larger amounts of 2-Fe/I, annealing experiments were conducted in which 2-Fe/H2O was stored at 75 °C, far below its decomposition temperature observed in the TG measurements. In this experiment, samples were taken at different times to monitor the decomposition process and were then measured by XRPD. Surprisingly, after 43 h, a mixture of the expected phase 2-Fe/I and an unknown crystalline phase was obtained (Fig. 2). By increasing the reaction time, the intensities of 2-Fe/I increased slightly, indicating a phase transition (Fig. S16†). Therefore, the experiment was repeated at slightly lower temperature and samples were taken at earlier times. This procedure leads to the new phase with only small traces of 2-Fe/I, indicating that this phase (2-Fe/II) has formed under kinetic control (Fig. 2).
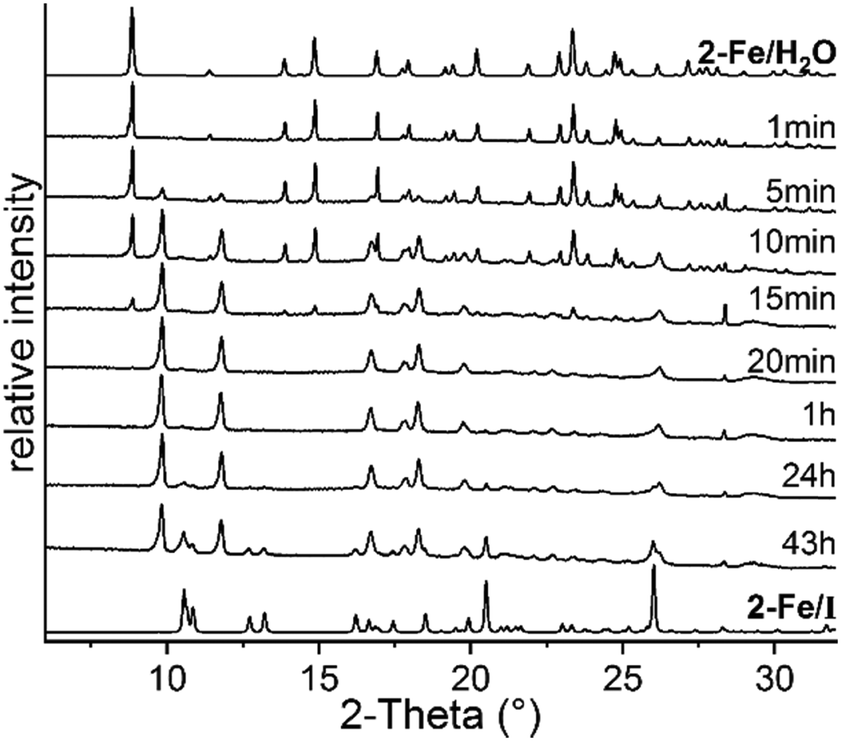 | ||
| Fig. 2 Ex situ XRPD pattern of samples obtained at different reaction times during heating of 2-Fe/H2O at 70 °C measured with Cu-Kα1 radiation together with the calculated pattern of 2-Fe/H2O and 2-Fe/I. | ||
The CN stretching mode for the new compound is observed at 2092 cm−1 in the IR and Raman spectra, almost identical to that of 2-Fe/I, indicating structural similarities (Fig. S17†). Later we found that heating 2-Fe/H2O for a few hours in a rotary evaporator at 50 °C and under 50 mbar leads to pure samples (Fig. S18†). Comparison of its powder pattern with those calculated for related thiocyanate coordination polymers with similar ligands reveals that the pattern of 2-Fe/II looks very similar to that of [Cu(NCS)2(4-cyanopyridine)2]n reported in the literature (Fig. S19†).59 Therefore, a Rietveld refinement was performed, which proves that both compounds are isotypic and that 2-Fe/II represents an isomer of 2-Fe/I (Fig. S20†).
To investigate the thermal behaviour of 1-Fe and 2-Fe/H2O in more detail, temperature dependent X-ray diffraction experiments were performed in open and closed capillaries. If 1-Fe is investigated in an open capillary, it decomposes at 130 °C without a transition into a crystalline phase (Fig. S21†). If this compound is heated in a sealed capillary, it transforms into 2-Fe/II (Fig. 3). In contrast, 2-Fe/H2O transforms into 2-Fe/II in an open and into 2-Fe/I in a closed capillary upon heating (Fig. 3). Surprisingly, no transition of 2-Fe/II into 2-Fe/I is observed as indicated by the annealing experiments (Fig. 2). Moreover, in none of these measurements a transformation into the crystalline phase (3-Fe) obtained after the second mass loss in the TG measurement of 1-Fe and 2-Fe/H2O is observed. However, as shown above, the new crystalline phase (3-Fe) can be obtained by thermal decomposition of 1-Fe or 2-Fe/H2O in a thermobalance.
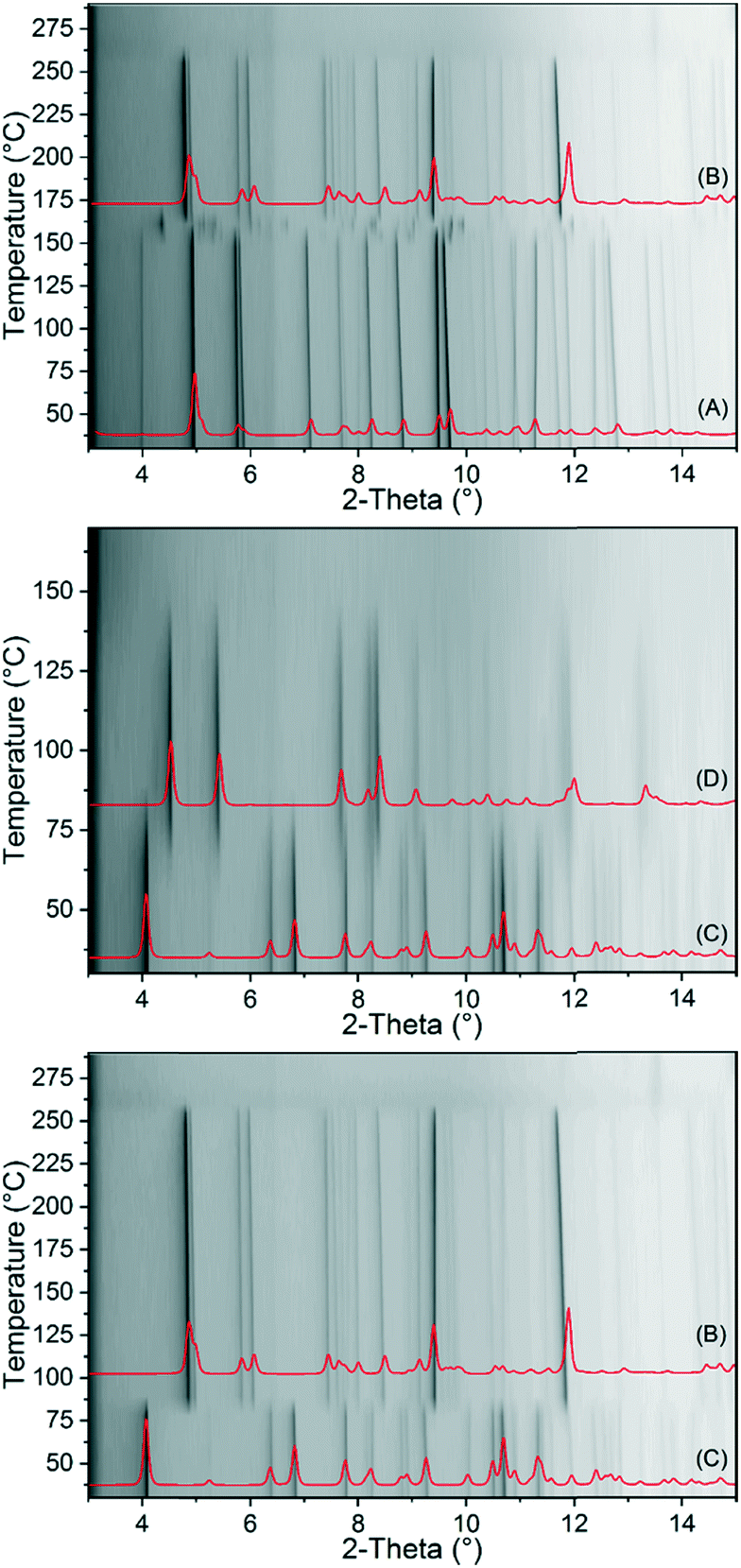 | ||
| Fig. 3 Temperature dependent XRPD patterns of 1-Fe in a sealed capillary (top) and of 2-Fe/H2O in an open (middle) and in a sealed capillary (bottom) measured with Cu-Kα1 radiation. The calculated patterns of 1-Fe (A), 2-Fe/I (B), 2-Fe/H2O (C) and 2-Fe/II (D) are given for comparison. | ||
Unfortunately, we have not found any known thiocyanate coordination polymer that might be isotypic to this phase. Its CN stretch is observed at 2122 cm−1 and at 2098 cm−1 in the Raman spectra, which indicates that a more condensed thiocyanate coordination polymer is obtained (Fig. S22†). Therefore, we investigated if such a compound can be prepared with cadmium, because, as mentioned above, this cation is much more chalcophilic and can easily form compounds with bridging thiocyanate anions. It is notable that two compounds with Cd(II) were already reported in the literature. This includes [Cd(NCS)2(4-picoline)2]n (2-Cd/I)57,58 which is isotypic to 2-Fe/I and [Cd(NCS)2(4-picoline)4·0.67 4-picoline·0.33 H2O (1-Cd).58,60,61 Therefore, to prove the existence of a compound with the composition [Cd(NCS)2(4-picoline)]n that might be isotypic to 3-Fe, both of them were synthesized and investigated by TG-DSC measurements. Both compounds were easily obtained and then investigated by IR and Raman spectroscopy as well as XRPD, which confirms the purity of the samples (Fig. S23–S26†). The TG curve of 1-Cd at a heating rate of 8 °C min−1 shows two mass steps of which the first one corresponds to the mass loss of two 4-picoline ligands and the solvent molecules, whereas in the second step two coligands are removed. There are no hints for the formation of a 4-picoline-deficient phase with the composition [Cd(NCS)2(4-picoline)]n but if 1-Cd is measured at a heating rate of 1 °C min−1 an additional mass step occurs, which becomes even more clear from the DSC and DTG curves since they are split (Fig. 4 and S27†). TG-DSC measurements for 2-Cd/I lead to comparable results (Fig. 4 and S28†).
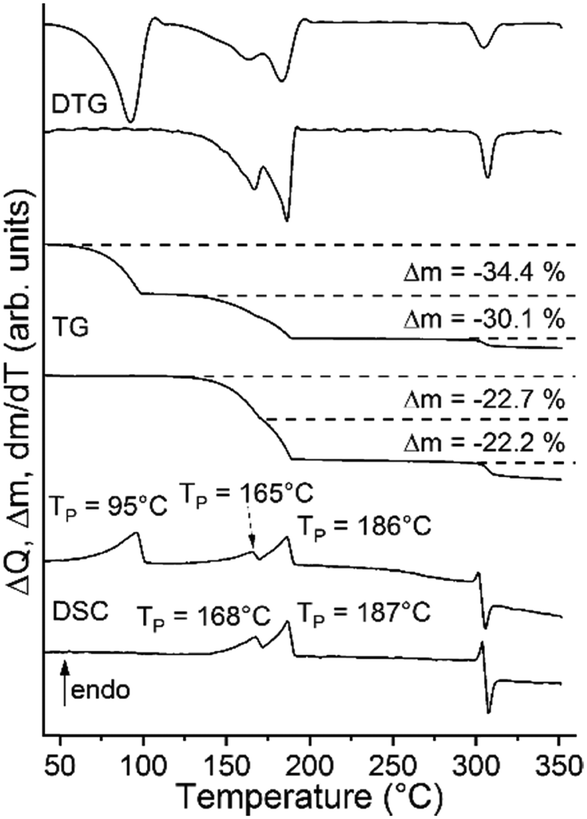 | ||
| Fig. 4 DSC, TG and DTG curves of 1-Cd (top curves) and 2-Cd/I (bottom curves) measured at a heating rate of 1 °C min−1. | ||
In further TG measurements the residues formed after the first mass loss of 2-Cd/I, respectively second mass loss of 1-Cd were isolated. The Raman spectrum of this residue is similar to that of 3-Fe and XRPD investigations reveal that it might be isotypic to 3-Fe (Fig. S29 and S30†). For completeness, 1-Cd was also investigated by temperature dependent XRPD measurements (Fig. 5).
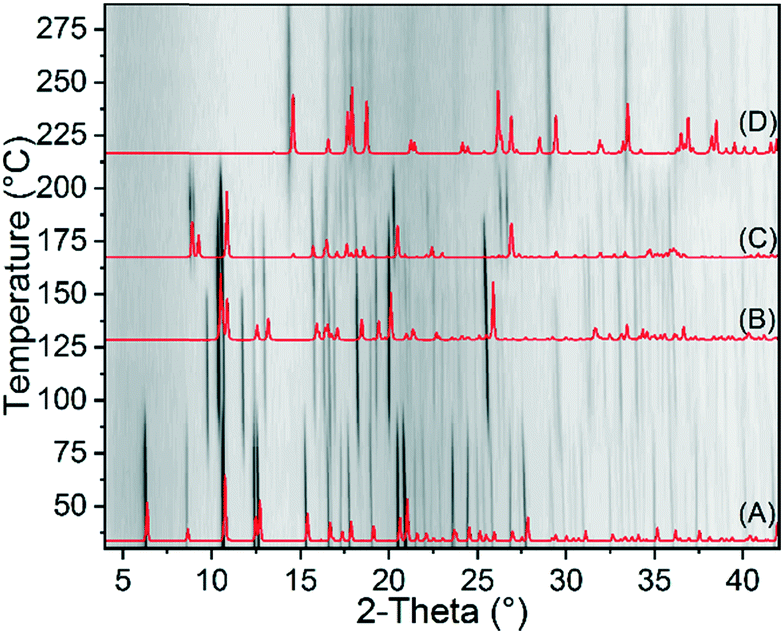 | ||
| Fig. 5 Temperature dependent XRPD pattern of 1-Cd measured in an open capillary with Cu-Kα1 radiation. The calculated patterns of 1-Cd (A), 2-Cd/I (B), 3-Cd (C) and Cd(NCS)2 (D) are given for comparison. | ||
In contrast to 1-Fe, 1-Cd transforms via2-Cd/I as an intermediate to 3-Cd. Surprisingly, there are some intense additional reflections which do not correspond to 2-Cd/I, indicating the formation of an additional crystalline phase (2-Cd/III, Fig. 5). Therefore, using slightly higher temperatures and different heating rates, additional TG measurements of 1-Cd were performed and the residues, isolated directly after the first mass loss, were investigated by XRPD (Fig. S31†). This shows that mixtures of 2-Cd/I and 2-Cd/III in different ratios are always obtained.
However, the TG measurements reveal that a Cd compound with the composition [Cd(NCS)2(4-picoline)]n (3-Cd) exists and that it should be isotypic to 3-Fe. There are also hints that a second modification of [Cd(NCS)2(4-picoline)2]n (2-Cd/I) can be obtained (2-Cd/III) but mostly mixtures with 2-Cd/I are formed. Therefore, we tried to synthesize these phases in solution. If Cd(NCS)2 and 4-picoline are reacted in acetonitrile at a ratio of 3:1, single crystals of 3-Cd were obtained. Its structure was determined by single crystal X-ray diffraction and by comparing the experimental with the calculated XRPD pattern using the single crystal data, it is obvious that a pure form was obtained (Fig. S32†). The crystallographic data of 3-Cd were used as a starting model to determine the crystal structure of 3-Fe by Rietveld refinements, which definitely proves that both forms are isotypic (Fig. S33†).
Additional crystallization experiments reveal that the new form 2-Cd/III is obtained by kinetic control from the reaction between Cd(NCS)2 and 4-picoline at a ratio of 1:1 (Fig. S34†). Unfortunately, no single crystals of this new form are available. However, its IR- and Raman spectra are very similar to those of 2-Cd/I, indicating that both structures are strongly related (Fig. S35†). The comparison of its powder pattern with that of related thiocyanate coordination compounds reveals similarities to that of [Cd(NCS)2(4-cyanopyridine)2]n already reported in the literature (Fig. S36†).62 Therefore, a Rietveld refinement was performed, which proves that both compounds are isotypic (Fig. S37†).
Crystal structures
2 -Fe/I is isotypic to 2-Cd/I as reported in the literature57,58 and crystallizes in the monoclinic space group C2/c with Z = 8 formula units in the unit cell. The asymmetric unit consists of two crystallographic independent metal cations that are octahedrally coordinated (Fig. 6).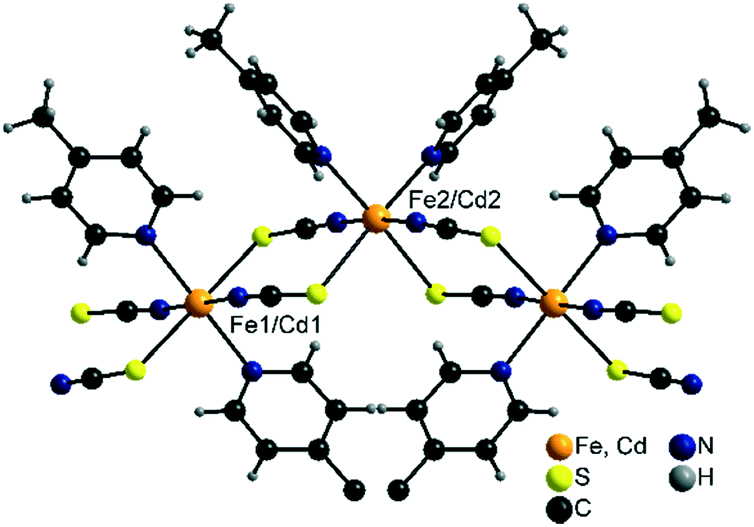 | ||
| Fig. 6 Part of a chain in the crystal structure of 2-Fe/I and 2-Cd/I. | ||
On one of these cations, the thiocyanate N and S atoms as well as the N atoms of the 4-picoline ligands are in trans-position, whereas on the other one, the thiocyanate S and the 4-picoline N atoms are cis (Fig. 6). The metal cations are linked into chains via pairs of μ-1,3-bridging thiocyanate anions which are corrugated because of the alternating trans- and cis–cis–trans coordination.
2 -Fe/II is isotypic to [Cu(NCS)2(4-cyanopyridine)2]n as reported in the literature.59 Both compounds crystallize in the monoclinic space group P21/c with Z = 4 formula units in the unit cell. In contrast to 2-Fe/I, all metal cations show the thiocyanate N and S atoms as well as the 4-picoline N atoms in trans position (Fig. 7). Consequently, metal cations are linked into linear chains by pairs of anionic ligands. Due to the different coordination environments and the different chain geometry compounds, 2-Fe/I and 2-Fe/II represent isomeric modifications.
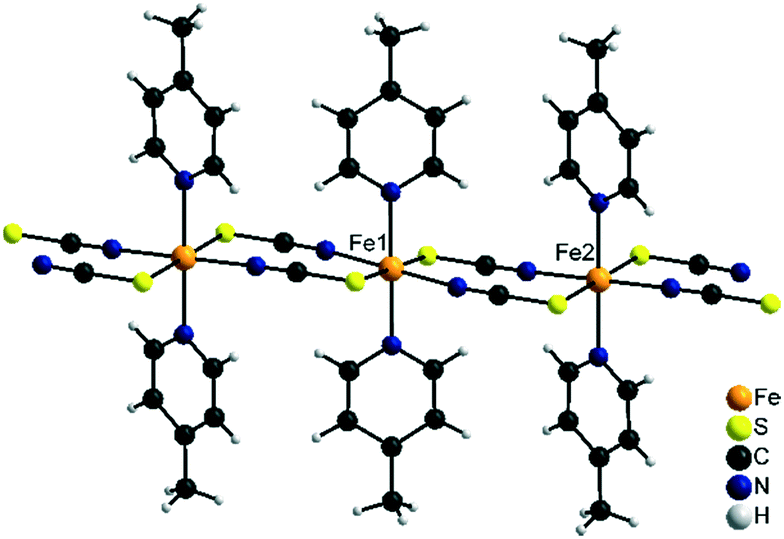 | ||
| Fig. 7 Part of a chain in the crystal structure of 2-Fe/II. | ||
Compound 2-Cd/III crystallizes in the monoclinic space group P21/n with Z = 2 and is isotypic to [Cd(NCS)2(4-cyanopyridine)2]n as reported in the literature.62 The Cd cations are located on centres of inversion and are octahedrally coordinated by two bridging N and two S coordinating thiocyanate atoms and two N atoms of the 4-picoline ligands (Fig. 8). As in the other chain compounds, the metal cations are linked into chains, which in this case are linear as observed in 2-Fe/II (Fig. 8).
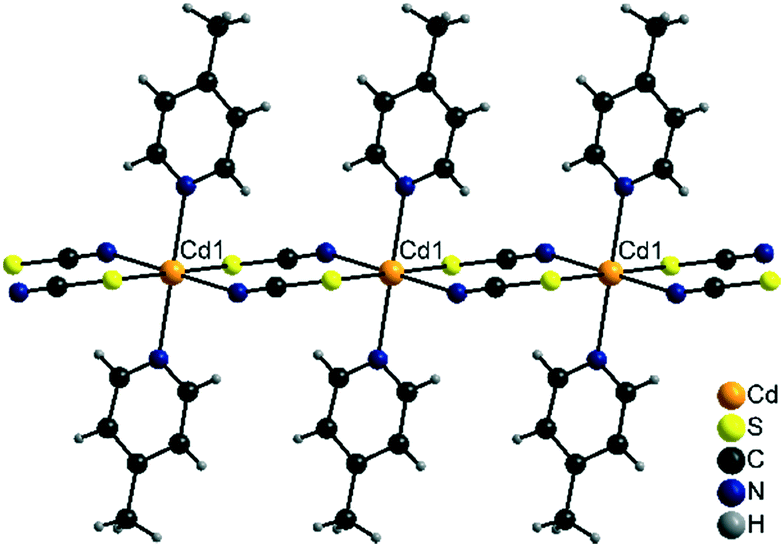 | ||
| Fig. 8 Part of a chain in the crystal structure of 2-Cd/III. | ||
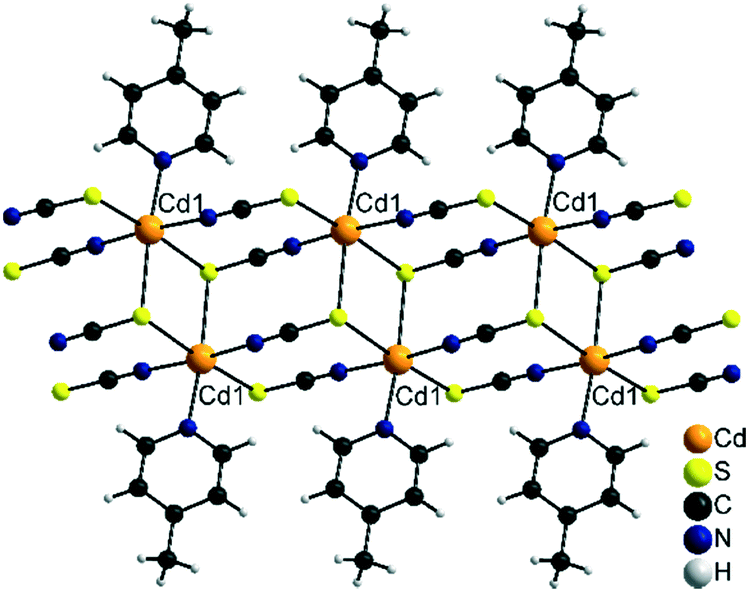 | ||
| Fig. 9 Part of a chain in the crystal structure of 3-Cd and the isotypic 3-Fe. | ||
As mentioned above, 2-Fe/II and 2-Cd/III are not isotypic and crystallize in different structure types. Differences between both structures can be predominantly found in the rotation of the 6-membered rings along the chains, which are nearly perpendicular in 2-Fe/II and parallel in 2-Cd/III (Fig. S38†). In contrast, the arrangement of the chains in the crystal structures of both compounds is very similar and reminiscent of a herringbone structure (Fig. S39†).
Finally, the most 4-picoline deficient compounds 3-Cd and 3-Fe crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2 and the metal cation in a general position. In their crystal structures the metal cations are octahedrally coordinated by three S and two N atoms of the thiocyanate anions and one N atom of the neutral co-ligand. The cations are linked by pairs of μ-1,3-bridging anionic ligands into linear chains. Two of these chains are interconnected by μ-1,3,3 (N, S, S) bridging thiocyanate anions into double chains (Fig. 9). It is notable that this is a very rare structural motif and only a very few examples, mostly with Cd(II) cations, are reported.49,63,64 With less chalcophilic metal cations, only one exception is known, which is [Ni(NCS)2(4-aminopyridine)]n.49 To the best of our knowledge, with Fe(NCS)2 such a chain structure was never observed before in thiocyanate chemistry.
with Z = 2 and the metal cation in a general position. In their crystal structures the metal cations are octahedrally coordinated by three S and two N atoms of the thiocyanate anions and one N atom of the neutral co-ligand. The cations are linked by pairs of μ-1,3-bridging anionic ligands into linear chains. Two of these chains are interconnected by μ-1,3,3 (N, S, S) bridging thiocyanate anions into double chains (Fig. 9). It is notable that this is a very rare structural motif and only a very few examples, mostly with Cd(II) cations, are reported.49,63,64 With less chalcophilic metal cations, only one exception is known, which is [Ni(NCS)2(4-aminopyridine)]n.49 To the best of our knowledge, with Fe(NCS)2 such a chain structure was never observed before in thiocyanate chemistry.
Thermodynamic relations
To investigate the thermodynamic relations of 2-Fe/I and 2-Fe/II as well as of 2-Cd/I and 2-Cd/III, solvent mediated conversion experiments were performed at room temperature. For this reason, suspensions of Fe and the respective Cd modifications were stirred in selected solvents. The residues, formed at different reaction times, were analysed by XRPD. If a mixture of 2-Fe/I and 2-Fe/II is investigated in acetonitrile/cyclohexane, the reflections of 2-Fe/II disappeared within 14 d, whereas those of 2-Fe/I are still present. However, an additional crystalline compound is formed as a by-product, which does not correspond to any of the known Fe phases (Fig. S40†). If this experiment is repeated in cyclohexane/ethanol, only a small amount of the unknown phase has formed and it is more obvious that 2-Fe/II transforms into 2-Fe/I (Fig. S41†). Similar experiments for 2-Cd/I and 2-Cd/III clearly show that in methanol, 2-Cd/III transforms into 2-Cd/I within 15 min (Fig. S42†). All this proves that both forms I represent the thermodynamically stable phases at room temperature. In this context, it is noted that the density of 2-Fe/I is higher than that of 2-Fe/II and that the density of 2-Cd/I is higher than that of 2-Cd/III, indicating that both forms I are also thermodynamically stable at absolute zero.65,66 To check if a solid–solid transformation between the different modifications can be observed and to determine the direction of the heat flow, DSC measurements were performed for 2-Fe/II and 2-Cd/III. Upon heating 2-Fe/II in a DSC, a weak exothermic signal is observed at about 125 °C followed by two poorly resolved endothermic events that might correspond to the formation of 3-Fe and its decomposition into Fe(NCS)2 (Fig. 10). Similar observations were made for 2-Cd/III even if the exothermic signal is weaker and shifted to higher temperatures (Fig. 10). To identify the intermediates of these reactions, the residues formed before and after the exothermic event and after the first endothermic signal were isolated and investigated by XRPD. As expected, the first endothermic signal corresponds to the removal of each 4-picoline ligand leading to the formation of 3-Fe and 3-Cd, respectively (Fig. 10 bottom and S43†). Surprisingly, the residue formed in the exothermic event corresponds to 2-Fe/I and 2-Cd/I, respectively. These results prove that a phase transition of 2-Fe/II into 2-Fe/I and of 2-Cd/III into 2-Cd/I has occurred.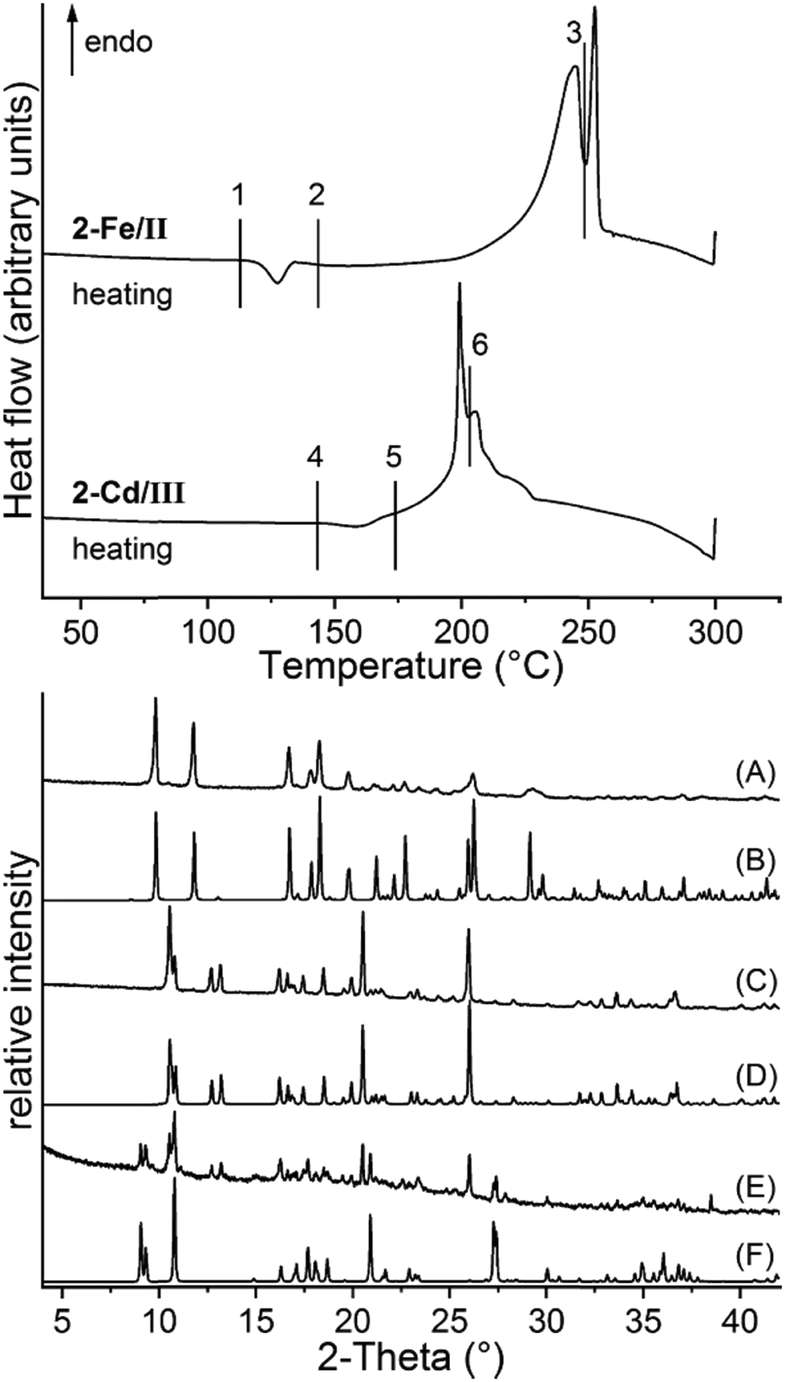 | ||
| Fig. 10 DSC measurements of 2-Fe/II and 2-Cd/III with heating rates of 10 °C min−1 together with markers where the products were isolated. The residues obtained at these points were investigated using XRPD. Shown are the powder pattern of the residues obtained at point 1 (A), 2 (C) and 3 (E) together with the calculated pattern of 2-Fe/II (B), 2-Fe/I (D) and 3-Fe (F). For the respective XRPD pattern for the Cd compounds, see Fig. S43.† | ||
Following the heat of transition rule of Burger and Ramberger,66 the exothermic signal indicates that both Fe and both Cd modifications are related by monotropism, with 2-Fe/I and 2-Cd/I thermodynamically stable over the whole temperature range and the corresponding energy–temperature diagrams can be found in Fig. S44.† In this context, it is notable that an exothermic phase transition is also expected for enantiotropic modifications but only if the phase transition takes place below the thermodynamic transition temperature. This, however, can be excluded because the density and the solvent mediated conversion experiments indicate that 2-Fe/I and 2-Cd/I are already thermodynamically stable at lower temperatures.
Similar experiments were also performed for 2-Fe/H2O and 1-Cd using heating rate dependent DSC measurements (Fig. S45 and S46†). For 2-Fe/H2O an endothermic peak is observed upon heating that corresponds to the formation of 2-Fe/II, and on further heating an exothermic peak is observed which corresponds to the phase transition of 2-Fe/II into 2-Fe/I (Fig. S47†). For 1-Cd no exothermic transition is observed, which might be traced back to the fact that in this case 2-Cd/I has formed by ligand removal.
Experimental
Materials and methods
Ba(NCS)2·3H2O was obtained from Alfa Aesar. FeCl2·4H2O, KSCN and CdSO4·8/3H2O were obtained from Merck. All chemicals were used without further purification. Cd(NCS)2 was prepared by the reaction of equimolar amounts of CdSO4·8/3H2O and Ba(NCS)2·3H2O in water. The resulting white precipitate of BaSO4 was filtered off and the filtrate was concentrated to complete dryness resulting in white residues of Cd(NCS)2. Fe(NCS)2 was always synthesized in situ by the reaction of FeCl2·4H2O with equimolar amounts of KSCN.Synthesis of Fe(NCS)2(4-picoline)4-solvate (1-Fe)
FeCl2·4H2O (1.0 mmol, 200 mg) and KSCN (2.0 mmol, 194 mg) were dissolved in 3.0 mL H2O. Then 3.0 mL of 4-picoline were added. Stirring was continued for around 5 minutes and the precipitate was then filtered off and dried in vacuum.Crystals of 1-Fe suitable for single crystal structure analysis were obtained by the reaction of FeCl2·4H2O (0.25 mmol, 49.7 mg) and KSCN (0.5 mmol, 48.6 mg) in 1.5 mL H2O with 4-picoline dissolved in 1.5 mL H2O (2.5 mmol, 243.3 μL). Elemental analysis yielded slightly varying results, which can be traced back to different amounts of solvents inside the channels of the sample.
Elemental analysis: calcd (%) for the solvate free complex C26H28FeN6S2 (544.53 g mol−1): C 57.35, H 5.18, N 15.43 and S 11.78; found: C 58.24, H 5.34, N 15.19 and S 10.4.
Synthesis of 2-Fe/H2O
FeCl2·4H2O (3.0 mmol, 600 mg) and KSCN (6.0 mmol, 576 mg) were dissolved in 20.0 mL H2O before adding 4-picoline (6.0 mmol, 6 × 97 μL). The precipitate was stirred for two minutes and then filtered off and left to dry. No crystals suitable for single X-ray structure analysis were obtained.Elemental analysis: calcd (%) for C14H18FeN4O2S2 (394.30 g mol−1): C 42.65, H 4.60, N 14.21 and S 16.26; found: C 42.64, H 4.60, N 14.27 and S 16.63.
Synthesis of 2-Fe/I
2 -Fe/I was obtained by thermal decomposition of 2-Fe/H2O only. Elemental analysis: calcd (%) for C14H14FeN4S2 (358.27 g mol−1): C 46.93, H 3.94, N 15.64 and S 17.90; found: C 46.66, H 3.89, N 15.31 and S 17.71.Synthesis of 2-Fe/II
2 -Fe/II was obtained by water removal from 2-Fe/H2O in a rotary evaporator at 45 °C and 40 mbar within a few hours. Elemental analysis: calcd (%) for C14H14FeN4S2 (358.27 g mol−1): C 46.93, H 3.94, N 15.64 and S 17.90; found: C 46.78, H 3.82, N 15.30 and S 17.70.Synthesis of 3-Fe
3 -Fe can only be obtained by thermal decomposition and is always contaminated by small amounts of 2-Fe/I and/or 2-Fe/II because the corresponding transition cannot successfully be resolved.Synthesis of [Cd(NCS)2(4-picoline)4] 0.67·4-picoline·0.33·H2O (1-Cd)
Cd(NCS)2 (1.0 mmol, 228.6 mg) was dissolved in water (2.0 mL) and an excess of 4-picoline (2.0 mL) was added to the solution. The product formed immediately; it was filtered off and washed with a 1:1 mixture of H2O and 4-picoline. Elemental analysis yielded varying results, presumably due to different amounts of solvents inside the channels of the sample.
Synthesis of [Cd(NCS)2(4-picoline)2]n (2-Cd/I)
Cd(NCS)2 (0.5 mmol, 114 mg) was dissolved in 2.0 mL H2O. To this solution 4-picoline (0.5 mmol, 48.3 μL) was added while stirring. The white powder that formed was stirred further for one day and then filtered off and dried in vacuum. Elemental analysis: calcd (%) for C14H14CdN4S2(414.82 g mol−1): C 40.54, H 3.4, N 13.51 and S 15.46; found: C 40.13, H 3.33, N 13.30 and S 15.21.Synthesis of [Cd(NCS)2(4-picoline)2]n (2-Cd/III)
Cd(NCS)2 (0.5 mmol, 114 mg) was suspended in 1.0 mL MeOH. The ligand 4-picoline (1.0 mmol, 97 μL) was added to the stirred suspension and the precipitate was filtered off almost immediately. It is noted that the product is sometimes contaminated with Cd(NCS)2 if prepared by this route.Synthesis of [Cd(NCS)2(4-picoline)]n (3-Cd)
Cd(NCS)2 (0.75 mmol, 171 mg) was stirred in 3.0 mL MeCN and 4-picoline (0.25 mmol, 24.3 μL) was added. The white precipitate was stirred further for one day and then filtered off and dried in vacuum. Elemental analysis: calcd (%) for C8H7CdN3S2 (321.70 g mol−1): C 29.87, H 2.19, N 13.06 and S 19.94; found: C 30.02, H 2.1, N 12.99 and S 20.36.Crystals of 3-Cd suitable for single crystal structure analysis were obtained by the reaction of Cd(NCS)2 (1.5 mmol, 343 mg) with 4-picoline (0.15 mmol, 14.5 μL) in MeCN (3.0 mL).
Single crystal structure analysis
Data collection was performed using an IPDS-1 for 3-Cd and an IPDS-2 for 1-Fe using Mo-Kα radiation. The structures were solved using direct methods with SHELXS-97 (ref. 67) and the structure refinement was performed against F2 using SHELXL-2014.68 For both compounds a numerical absorption correction was performed using X-Red and X-Shape of the software package X-Area. All non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms were positioned with idealized geometry (methyl H atoms were allowed to rotate but not to tip) and were refined isotropically with Uiso (H) = −1.2 Ueq (C) (1.5 for methyl H) using a riding model. Selected crystal data are given in Table S1 in the ESI.† Compound 1-Fe contains a very small amount of disordered solvent in channels of the structure, for which no reasonable structure model can be found. Therefore, the data were corrected for disordered solvent using Squeeze in Platon. In 3-Cd the 4-picoline coligand is disordered and was refined using a split model. Selected crystal data and results of the structure refinements can be found in Table S1.†CCDC-1957533 (3-Cd) and CCDC-1957534 (1-Fe) contain the supplementary crystallographic data for this paper.
IR and Raman spectroscopy
IR spectra were recorded at RT on a Bruker Vertex70 FT-IR spectrometer using a broadband spectral range extension VERTEX FM for full mid and far IR. Raman spectra were recorded at RT on a Bruker RAM II FT-Raman spectrometer using a liquid nitrogen cooled, highly sensitive Ge detector, 1064 nm radiation and 3 cm−1 resolution.Thermogravimetry and differential scanning calorimetry
The TG-DSC measurements were performed on a STA-PT1000 and STA-PT1600 thermobalance from Linseis. Measurements were performed in Al2O3 crucibles and under a dynamic nitrogen atmosphere. The instruments were calibrated using standard referencing materials and corrected for buoyancy.Differential scanning calorimetry
The DSC experiments were performed using a DSC 1 star system with STARe Excellence software from Mettler-Toledo AG under a dynamic nitrogen flow in Al pans.Elemental analysis
For the elemental analysis a vario MICRO cube from Elementar Analysensysteme GmbH was used.XRPD structure determination
Structure solution and refinement of the crystal structure of 2-Fe/H2O were performed with the program TOPAS 6.0.69 Indexing was carried out by an iterative use of singular value decomposition (LSI) as implemented in TOPAS 6.0.70 The peak profile was determined by Pawley refinement71 using the fundamental parameter approach implemented into TOPAS (ref. 72) and the background was modelled using Chebyshev polynomials of 8th order. The hump in the background of the diffraction pattern caused by the glass capillary and a small amount of amorphous phase was modelled with very broad Lorentzian-type peaks. The crystal structure was determined by applying the global optimization method of simulated annealing (SA)73 in real space. The thiocyanate and 4-picoline molecules were described using rigid bodies in z-matrix notation and their rotation and translation modes were allowed to refine. The bond lengths and angles were taken from related single crystal structure data. The simulated annealing process was carried out iteratively. The positions of the iron atoms and all ligands were gradually fixed during the simulated annealing process once the coordination geometry was reasonable. Eventually, the background, lattice parameters, translations and the atomic positions of the iron atoms were refined without any constraints during the final Rietveld refinement.52 All hydrogen atoms were fixed at geometric calculated positions and distinct isotropic displacement parameters were applied for iron, thiocyanate, 4-picoline and water molecules. The final agreement factors are listed in Table S2 in the ESI.† The fit of the whole powder pattern is shown in Fig. S5 in the ESI.†CCDC-1957535 (2-Fe/H2O), CCDC-1957536 (2-Fe/I), CCDC-1957537 (3-Fe), CCDC-1957538 (2-Cd/III), CCDC-1957539 (2-Fe/II) contain the supplementary crystallographic data for this paper.
Laboratory X-ray powder diffraction (XRPD)
XRPD measurements for identification of the samples and for Rietveld refinement of 2-Cd/III were carried out at room temperature on a laboratory powder diffractometer in Debye–Scherrer geometry (Stadi P-Diffractometer (Stoe), Cu-Kα1 radiation from a primary Ge(111)-Johann-type monochromator, 1 Mythen 1 K detector (Dectris)). XRPD measurements of 2-Fe/H2O, 2-Fe/I, 2-Fe/II and 3-Fe were carried out at room temperature on a laboratory powder diffractometer in Debye–Scherrer geometry (Stadi P-Diffractometer (Stoe), Mo-Kα1 radiation from a primary Ge(111)-Johann-type monochromator, array of 3 Mythen 1 K detectors (Dectris)). All samples were loaded into 0.5 mm diameter glass capillaries (WJM-Glas/Mueller GmbH), which were spun during the measurements. The pattern of 2-Cd/III was measured in the 2θ range from 3.0° to 80.0° applying a total scan time of 6 hours per measurement whereas the patterns of 2-Fe/H2O, 2-Fe/I, 2-Fe/II and 3-Fe were measured using one detector in the 2θ range from 2° to 50° applying a total scan time of 16 hours per measurement.Rietveld refinements
Rietveld refinements52 of the crystal structure of 2-Cd/III, 2-Fe/I, 3-Fe/II and 3-Fe were performed using the program TOPAS 6.0.69 The profile function was described with the fundamental parameter approach implemented into TOPAS72 and the background was modelled using Chebyshev polynomials. The hump in the background of the diffraction pattern caused by the glass capillary was modelled with very broad Lorentzian type peaks. The thiocyanate and 4-picoline molecules were described using rigid bodies in z-matrix notation and their rotation and translation modes were allowed to refine. Bond lengths and angles were taken from isotypic single crystal structural data from the literature58,59,62 or from single crystal data presented in this paper. All hydrogen atoms were fixed at geometric calculated positions except in 3-Fe where no hydrogen atoms were added on the methyl group of 4-picoline because of the rotational disorder of the ligand. Distinct isotropic displacement parameters were applied for metals, thiocyanate and 4-picoline. Eventually, the background, lattice parameters, translations of rigid bodies and the atomic positions of cadmium and iron atoms were refined without any constraints during the final Rietveld refinement. The final agreement factors are listed in Table S2.† The fits of the whole powder patterns are shown in Fig. S14 (2-Fe/I), S19 (2-Fe/II), S32 (3-Fe) and S36 (2-Cd/III) in the ESI.†In situ X-ray powder diffraction
Temperature dependent in situ XRPD measurements were carried out on two laboratory powder diffractometers in Debye–Scherrer geometry: Bruker D8 Advance with Mo-Kα1 radiation from a primary Ge(110) Johansson-type monochromator and a LYNXEYE detector (for 1-Fe and 2-Fe/H2O) and Bruker D8 Advance with Cu-Kα1 radiation from a primary Ge(110) Johansson-type monochromator and Våntec detector (for 1-Cd). The samples were loaded into 0.5 mm diameter quartz capillaries, which were spun during the measurements. The patterns were measured in the 2θ range from 2.0° to 60.0° for 1-Cd, from 2.0° to 30.0° for 2-Fe/H2O, from 3.0° to 20.0° for 1-Fe (with open capillary) and from 3.0 to 15.0° for 1-Fe (with sealed capillary). A total scan time of 3 hours (for 1-Cd and 2-Fe/H2O) and 4 hours (for 1-Fe) was applied per each measurement. The temperature was adjusted using a TC-transmission furnace (WJM-Glas/Mueller GmbH). 1-Cd was heated from 30 °C to 130 °C in 10 °C steps and from 130 °C to 250 °C in 5 °C steps at a heating rate of 2 °C min−1. 2-Fe/H2O was heated from 30 °C to 160 °C in 10 °C steps and from 160 °C to 250 °C in 5 °C steps at a heating rate of 2 °C min−1. 1-Fe was heated from 30 °C to 130 °C in 10 °C steps and from 130 °C to 250 °C in 5 °C steps at a heating rate of 2 °C min−1. During each step, a diffraction pattern was collected after a delay time of 5 minutes to ensure thermal equilibration of the sample.Conclusions
In this contribution a variety of new coordination compounds based on Fe(NCS)2 and Cd(NCS)2 were reported. With Fe(II) only discrete complexes can be obtained from solution, whereas the corresponding coordination polymers can only be prepared by thermal annealing in the solid state. More importantly, careful analysis of their thermal properties leads to the discovery of two new isomers and one additional chain compound of unusual geometry, which for Fe(II) was never observed before. All this demonstrates the importance of this approach for the synthesis of new coordination compounds that are not accessible from the liquid state. This approach, however, leads only to the formation of microcrystalline powders but in this case the corresponding more chalcophilic Cd(II) compounds can be prepared to retrieve structural information on their Fe(II) analogs. Alternatively, one can search for similar compounds that even if they contain different co-ligands might be isotypic. We have also shown that careful analysis of the thermal behaviour of the isomers of [Fe(NCS)2(4-picoline)2]n (2-Fe) and [Cd(NCS)2(4-picoline)2]n (2-Cd) allows determination of their thermodynamic relation, which is important information concerning the stability of stable and metastable phases. In this context, it is noted that it is not in contradiction that the solid state transition was observed in DSC but not in the temperature dependent XRPD measurements. Such transitions proceed by nucleation and growth of the new phase for which some nucleation energy is required. Therefore, such transitions can be suppressed but also initiated if, e.g., a few nuclei of the stable phase are present.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Deutsche Forschungsgemeinschaft (Project No. Na 720/5-2) and the State of Schleswig-Holstein. We thank Prof. Dr. W. Bensch for providing access to his experimental facilities. Open Access funding provided by the Max Planck Society.References
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed
.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martínez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-García and M. Julve, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed
- D. Braga, M. Curzi, F. Grepioni and M. Polito, Chem. Commun., 2005, 2915–2917, 10.1039/b503404c
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
- D. Braga, S. L. Giaffreda, F. Grepioni, A. Pettersen, L. Maini, M. Curzi and M. Polito, Dalton Trans., 2006, 1249–1263 RSC
- C. Näther, S. Wöhlert, J. Boeckmann, M. Wriedt and I. Jess, Z. Anorg. Allg. Chem., 2013, 639, 2696–2714 CrossRef
- A. Zurawski, J. C. Rybak, L. V. Meyer, P. R. Matthes, V. Stepanenko, N. Dannenbauer, F. Würthner and K. Müller-Buschbaum, Dalton Trans., 2012, 41, 4067–4078 RSC
- C. Näther, G. Bhosekar and I. Jess, Inorg. Chem., 2007, 46, 8079–8087 CrossRef PubMed
- C. J. Höller and K. Müller-Buschbaum, Inorg. Chem., 2008, 47, 10141–10149 CrossRef PubMed
- S. Suckert, M. Rams, L. Germann, D. M. Cegiełka, R. E. Dinnebier and C. Näther, Cryst. Growth Des., 2017, 17, 3997–4005 CrossRef CAS
- K. Müller-Buschbaum, Z. Anorg. Allg. Chem., 2005, 631, 811–828 CrossRef
- C. J. Adams, P. C. Crawford, A. G. Orpen, T. J. Podesta and B. Salt, Chem. Commun., 2005, 2457–2458, 10.1039/b501555c
- C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790–1795 RSC
- T. Den, P. M. Usov, J. Kim, D. Hashizume, H. Ohtsu and M. Kawano, Chem. – Eur. J., 2019, 25, 11512–11520 CrossRef CAS PubMed
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed
- T. Stolar, L. Batzdorf, S. Lukin, D. Žilić, C. Motillo, T. Friščić, F. Emmerling, I. Halasz and K. Užarević, Inorg. Chem., 2017, 56, 6599–6608 CrossRef CAS PubMed
- A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Chem., 2015, 6, 6662 CAS
- S. Wöhlert, T. Runčevski, R. E. Dinnebier, S. G. Ebbinghaus and C. Näther, Cryst. Growth Des., 2014, 14, 1902–1913 CrossRef
- J. Werner, T. Runčevski, R. Dinnebier, S. G. Ebbinghaus, S. Suckert and C. Näther, Eur. J. Inorg. Chem., 2015, 2015, 3236–3245 CrossRef CAS
- T. Neumann, M. Rams, Z. Tomkowicz, I. Jess and C. Näther, Chem. Commun., 2019, 55, 2652–2655 RSC
- K. Akhbari and A. Morsali, Inorg. Chim. Acta, 2015, 429, 109–113 CrossRef CAS
- H. Ohtsu and M. Kawano, Chem. Commun., 2017, 53, 8818–8829 RSC
- S. Suckert, M. Rams, M. M. Rams and C. Näther, Inorg. Chem., 2017, 56, 8007–8017 CrossRef CAS PubMed
- C. Näther and I. Jess, Eur. J. Inorg. Chem., 2004, 2868–2876 CrossRef
- S. S. Massoud, M. Dubin, A. E. Guilbeau, M. Spell, R. Vicente, P. Wilfling, R. C. Fischer and F. A. Mautner, Polyhedron, 2014, 78, 135–140 CrossRef CAS
- F. A. Mautner, M. Scherzer, C. Berger, R. C. Fischer, R. Vicente and S. S. Massoud, Polyhedron, 2015, 85, 20–26 CrossRef CAS
- F. A. Mautner, R. C. Fischer, L. G. Rashmawi, F. R. Louka and S. S. Massoud, Polyhedron, 2017, 124, 237–242 CrossRef CAS
- C. J. Adams, J. A. Real and R. E. Waddington, CrystEngComm, 2010, 12, 3547–3553 RSC
- J. G. Małecki, T. Groń and H. Duda, Polyhedron, 2012, 36, 56–68 CrossRef
- B. Machura, J. Palion, M. Penkala, T. Groń, H. Duda and R. Kruszynski, Polyhedron, 2013, 56, 189–199 CrossRef CAS
- Y. P. Prananto, A. Urbatsch, B. Moubaraki, K. S. Murray, D. R. Turner, G. B. Deacon and R. Batten Stuart, Aust. J. Chem., 2017, 70, 516–528 CrossRef CAS
- F. A. Mautner, M. Traber, R. C. Fischer, A. Torvisco, K. Reichmann, S. Speed, R. Vicente and S. S. Massoud, Polyhedron, 2018, 154, 436–442 CrossRef CAS
- J. Palion-Gazda, B. Machura, F. Lloret and M. Julve, Cryst. Growth Des., 2015, 15, 2380–2388 CrossRef CAS
- M. Rams, Z. Tomkowicz, M. Böhme, W. Plass, S. Suckert, J. Werner, I. Jess and C. Näther, Phys. Chem. Chem. Phys., 2017, 19, 3232–3243 RSC
- S. Wöhlert, T. Fic, Z. Tomkowicz, S. G. Ebbinghaus, M. Rams, W. Haase and C. Näther, Inorg. Chem., 2013, 52, 12947–12957 CrossRef PubMed
- S. Wöhlert, Z. Tomkowicz, M. Rams, S. G. Ebbinghaus, L. Fink, M. U. Schmidt and C. Näther, Inorg. Chem., 2014, 53, 8298–8310 CrossRef PubMed
- J. L. Guillet, I. Bhowmick, M. P. Shores, C. J. A. Daley, M. Gembicky, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2016, 55, 8099–8109 CrossRef CAS PubMed
- C. Wellm, M. Rams, T. Neumann, M. Ceglarska and C. Näther, Cryst. Growth Des., 2018, 18, 3117–3123 CrossRef CAS
- R. González, A. Acosta, R. Chiozzone, C. Kremer, D. Armentano, G. De Munno, M. Julve, F. Lloret and J. Faus, Inorg. Chem., 2012, 51, 5737–5747 CrossRef PubMed
- J. Palion-Gazda, I. Gryca, A. Maroń, B. Machura and R. Kruszynski, Polyhedron, 2017, 135, 109–120 CrossRef CAS
- F. A. Mautner, C. Berger, R. C. Fischer and S. S. Massoud, Inorg. Chim. Acta, 2016, 448, 34–41 CrossRef CAS
- J. Werner, M. Rams, Z. Tomkowicz and C. Näther, Dalton Trans., 2014, 43, 17333–17342 RSC
- J. Werner, Z. Tomkowicz, M. Rams, S. G. Ebbinghaus, T. Neumann and C. Näther, Dalton Trans., 2015, 44, 14149–14158 RSC
- S. Suckert, M. Rams, M. Böhme, L. S. Germann, R. E. Dinnebier, W. Plass, J. Werner and C. Näther, Dalton Trans., 2016, 45, 18190–18201 RSC
- T. Neumann, M. Ceglarska, M. Rams, L. S. Germann, R. E. Dinnebier, S. Suckert, I. Jess and C. Näther, Inorg. Chem., 2018, 57, 3305–3314 CrossRef CAS PubMed
- T. Neumann, I. Jess, F. Pielnhofer and C. Näther, Eur. J. Inorg. Chem., 2018, 4972–4981 CrossRef CAS
- T. Neumann, I. Jess, L. S. Germann, R. E. Dinnebier and C. Näther, Cryst. Growth Des., 2019, 19, 1134–1143 CrossRef CAS
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS
- J. D. Harris, W. E. Eckles, A. F. Hepp, S. A. Duraj, P. E. Fanwick, J. Richardson and E. M. Gordon, Mater. Des., 2001, 22, 625–634 CrossRef CAS
- J. D. Harris, W. E. Eckles, A. F. Hepp, S. A. Duraj and P. E. Fanwick, Inorg. Chim. Acta, 2002, 338, 99–104 CrossRef CAS
- C. W. Frank and L. B. Rogers, Inorg. Chem., 1966, 5, 615–622 CrossRef CAS
- R. A. Bailey, S. L. Kozak, T. W. Michelsen and W. N. Mills, Coord. Chem. Rev., 1971, 6, 407–445 CrossRef CAS
- M. Taniguchi, M. Shimoi and A. Ouchi, Bull. Chem. Soc. Jpn., 1986, 59, 2299–2302 CrossRef CAS
- R. Marsh, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 897–907 CrossRef
- B. Machura, A. Świtlicka, J. Mroziński, B. Kalińska and R. Kruszynski, Polyhedron, 2013, 52, 1276–1286 CrossRef CAS
- Y. A. Dyadin, N. V. Kislykh, G. N. Chekhova, N. V. Podberezskaya, N. V. Pervukhina, V. A. Logvinenko and I. M. Oglezneva, J. Inclusion Phenom., 1984, 2, 333–340 CrossRef CAS
- N. V. Pervukhina, N. V. Podberezskaya, V. V. Bakakin, N. V. Kislykh, G. N. Chekhova and Y. A. Dyadin, J. Struct. Chem., 1985, 26, 934–941 CrossRef
- C. Wanzhi, L. Fenghui and Y. Xiaozeng, Bull. Chem. Soc. Jpn., 2002, 75, 1559–1560 CrossRef
- S. Wöhlert, I. Jess and C. Näther, Z. Anorg. Allg. Chem., 2013, 639, 385–391 CrossRef
- S. Wöhlert, J. Boeckmann, I. Jess and C. Näther, CrystEngComm, 2012, 14, 5412–5420 RSC
-
A. I. Kitaigorodski, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 259–271 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
-
A. A. Coelho, Topas 6.0, Bruker AXS, Karlsruhe, 2017 Search PubMed
- A. A. Coelho, J. Appl. Crystallogr., 2003, 36, 86–95 CrossRef CAS
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS
- R. W. Cheary, A. Coelho and J. P. Cline, J. Res. Natl. Inst. Stand. Technol., 2004, 109, 1–25 CrossRef CAS PubMed
- A. Coelho, J. Appl. Crystallogr., 2000, 33, 899–908 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1957533–1957539. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01589b |
| This journal is © The Royal Society of Chemistry 2020 |