
Cu-Deficient plasmonic Cu2−xS nanocrystals induced tunable photocatalytic activities†
Xiao
Shao
a,
Tianyong
Zhang
*abc,
Bin
Li
 *ac,
Yue
Wu
a,
Xiaoyuan
Ma
a,
Jingchao
Wang
a and
Shuang
Jiang
*a
*ac,
Yue
Wu
a,
Xiaoyuan
Ma
a,
Jingchao
Wang
a and
Shuang
Jiang
*a
aTianjin Key Laboratory of Applied Catalysis Science and Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300354, China. E-mail: tyzhang@tju.edu.cn; libin@tju.edu.cn; shuangjiang@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
cTianjin Engineering Research Center of Functional Fine Chemicals, Tianjin 300354, China
First published on 30th November 2019
Abstract
Copper sulfide (Cu2−xS) is a kind of cation deficient transition metal sulfide, which has attracted great interest due to its unique properties that arise from degenerate vacancy-doping, particularly its tunable localized surface plasmon resonance (LSPR) properties. In this work, a range of vacancy-doped Cu2−xS nanocrystals (NCs) (Cu1.2S, Cu1.4S, Cu1.75S, and Cu1.94S NCs) with a size of around 10 nm was prepared with tunable LSPR through a hot injection method. The doping level was manipulated effectively by varying the injection volume of sulfur powder–oleic acid (S–OA) to finely tune the LSPR wavelength. Cu2−xS NCs with four different doping levels were investigated as photocatalysts for degradation of dyes. The Cu1.94S NCs with the highest LSPR energy exhibited the best photocatalytic activity due to the highest free carrier density, presumably influenced by Cu vacancies and the volume of the S–OA solution. Theoretical calculations of the free carrier density of Cu2−xS NCs are consistent with the order of the photocatalytic activity. The results demonstrate that the correlation of the photocatalytic activity with the corresponding concentration of free holes and the LSPR wavelength is of interest, which will provide inspiration for the design of non-noble metal catalysts by regulating the free hole concentration in degenerate p-doped Cu2−xS caused by cation vacancies.
Introduction
Copper chalcogenide Cu2−xE (E = S, Se or Te and x from 1 to 2) nanomaterials have attracted great interest and have been considered as potential materials for application in photocatalysis,1 photothermal therapy,2,3 photovoltaics,4 sensing,5 optical filters, superionic materials1–3 and nanoplasmonics.6–9 They have been found to show localized surface plasmon resonance (LSPR) behavior which was only reported in noble metals like Au, Ag and Cu nanomaterials previously. LSPR is the collective oscillation of free holes caused by copper deficiency.7 The resonance between the charge carrier oscillation and the incident light frequency produces a strong extinction band at the near infrared (NIR) wavelength. The peak LSPR extinction wavelength in the copper chalcogenide nanocrystal (NC) depends on the concentration of free holes directly related to the stoichiometry of the NC. By varying the Cu stoichiometry of the nanostructures, the LSPR excited wavelength is tunable within the infrared range.6,10In recent years, considerable attention has been paid to the environmental problem of organic pollutants in water. Among them, dyes constitute a large family and are a major threat to water pollution.7 Photocatalysis, especially with semiconductor materials as photocatalysts, has proved to be an effective technology to degrade organic contaminants and has received considerable attention.11–15 Cu2−xS nanomaterials display a bulk bandgap of 1.1–1.4 eV, varying with different stoichiometries of Cu to S.16 Therefore, Cu2−xS semiconductors are often used as photocatalysts because of their narrow bandgap. It has been reported in many studies that morphologically different Cu2−xS microstructures could be utilized as photocatalysts to destroy or decompose organic pollutants in water.1,17–20 Notably, copper sulphide nanomaterials, with non-toxic, inexpensive, stable and excellent catalytic properties, would be ideal to use in purification techniques.8
However, to the best of our knowledge, the photocatalytic activity of vacancy-doped Cu2−xS at the nanoscale, especially nanoparticles with a diameter below 20 nm have not yet been explored. Cu2−xS catalysts at the nanoscale are superior with a high specific surface area, resulting in more reactive sites and more unsaturated surface coordination sites exposed to the reactants.21 Besides, it is feasible to regulate the carrier concentrations through controlling the Cu/S feed ratio.22 Catalysts with a high specific surface area can provide abundant active sites for the adsorption of reactant molecules, which renders the catalytic process more efficient.21 The modulation from the micron level to the nanometer level of copper sulfide photocatalysts needs to be carried out, which will provide the possibility for the scale span of photocatalysts in the future. Besides, since LSPR depends on the carrier concentration of NCs, it can be reasonably assumed that the catalytic activity of NCs is related to their LSPR properties. Herein, a series of Cu2−xS NCs with different sizes have been prepared to investigate the photocatalytic behavior.
In this work, a series of monodisperse Cu2−xS NCs (Cu1.2S, Cu1.4S, Cu1.75S, Cu1.94S) have been successfully obtained with tunable LSPR through a hot-injection approach. The synthesis is tunable to optimize the photocatalytic properties of the NCs through varying the injection volume of sulfur powder–oleic acid (S–OA). The band structures of various possible crystal phases of Cu2−xS exhibit stoichiometry-dependent Fermi levels and bandgaps. During the synthesis of Cu2−xS NCs, we pay attention to the inherent correlations among elemental composition, morphology, and optical properties. The LSPR energy is strongly influenced by the injection volume of sulfur powder–oleic acid (S–OA) used during the hot injection. In this study, photocatalytic degradation of methyl orange (MO) and rhodamine B (RhB) was carried out with four kinds of Cu2−xS NCs as photocatalysts under UV light irradiation. As a result, we found that the photocatalytic activity could be regulated by monitoring the x value of Cu2−xS NCs on account of tunable carrier concentrations.
Experimental section
Materials and methods
Copper(I) chloride (CuCl, 99.95%), oleylamine (OM, 95%) and oleic acid (OA, AR) were purchased from Aladdin Reagent Company. Sulfur powder (S, 99.5%) was purchased from Sigma Aldrich. Methyl orange (AR, denoted as MO), rhodamine B (AR, denoted as RhB) were purchased from Tianjin Yuanli Chemical Technology Co. Ltd. The solvents used in all experiments or tests, including ethanol, trichloroethylene (TCE, refractive index (R = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 767)), chloroform (R = 1.4458), hexane (R = 1.375), cyclohexane (R = 1.4264) and toluene (R = 1.4969) were obtained from Tianjin Chemical Reagent, China. Water throughout all experiments was obtained via a Milli-Q water system. All of the chemicals were used as received without further purification.
767)), chloroform (R = 1.4458), hexane (R = 1.375), cyclohexane (R = 1.4264) and toluene (R = 1.4969) were obtained from Tianjin Chemical Reagent, China. Water throughout all experiments was obtained via a Milli-Q water system. All of the chemicals were used as received without further purification.
Synthesis of Cu2−xS NCs
Cu2−xS NCs were prepared using a modified procedure described by Zhu et al.22 For a typical synthetic reaction, air-free conditions accomplished by a standard Schlenk-line technique is necessary for all procedures carried out in this work. The S–OA precursor solution was prepared by dissolving 0.128 g (4 mmol) of S powder to 24 mL of OA in a 50 mL three-neck flask, and then the mixture was heated to 120 °C. At the same time, the Cu-OM precursor solution was prepared by adding 0.396 g (4 mmol) CuCl to 40 mL of OM in a 100 mL four-neck flask, and then the mixture was heated to 190 °C to obtain a yellow clear solution. Afterwards, the resultant clear yellow solution was cooled to 120 °C, followed by the quick injection of the S–OA solution into the Cu–OM solution. With the color of the solution changing to black from yellow, Cu2−xS NCs were formed. Subsequently, the reaction temperature was kept at 120 °C for 5 min to ensure that the growth conditions of Cu2−xS NCs could be satisfied. After the reaction completed, the mixture was cooled to room temperature by placing the four-neck flask in cool water. After adding excess ethanol into the solution and centrifugation at 6000 rpm for 10 min, the sediment of NCs was obtained. Chloroform was used to redisperse the precipitate, followed by adding excess ethanol and centrifuging at 6000 rpm for another 5 min. The above experimental procedure was repeated three times to obtain pure NCs. Finally, the as-obtained NCs were dried in a vacuum or dispersed in TCE for further characterization. Different volumes (21, 17, 14 and 12 mL) of the S–OA solution were injected into the Cu-OM solution to form Cu1.2S, Cu1.4S, Cu1.75S and Cu1.94S NCs, respectively.Photocatalytic activity measurement of Cu2−xS NCs
The photocatalytic performance of the Cu2−xS NCs was mainly measured by degrading MO and RhB solutions. The concentration of all degradation product solutions was 10 mg L−1. A 500 W high-pressure mercury lamp was used to generate UV light. All experiments were carried out at a temperature of 25 ± 2 °C. Typically, a 3 × 10−3 mmol Cu2−xS (about 50 mg Cu1.94S) catalyst was added into 100 ml of aqueous solution under ultrasonic irradiation to form a suspension, which was then magnetically stirred in the dark for 60 min to establish the adsorption–desorption equilibrium. The solution was collected after the light illumination every 20 min. Then the substrate concentrations were tested using a UV–vis spectrophotometer at corresponding wavelengths.Characterization
Results and discussion
Morphological, structural, and optical analyses of the Cu2−xS nanostructures
In this work, four kinds of high dispersive Cu2−xS NCs were synthesized through a hot injection method. Compared to previous reports, we greatly shorten the reaction time from 2 hours to 5 min, and the size distribution of NCs is narrowed.22 TEM techniques were adopted to characterize the size and morphology of the as-obtained Cu2−xS NCs. Fig. 1a–d show the TEM images of Cu1.2S, Cu1.4S, Cu1.75S and Cu1.94S NCs, respectively, obtained by varying the injection volumes (21, 17, 14 and 12 mL) of the S–OA solution with an injection temperature of 120 °C. As shown in Fig. 1a–c, the Cu2−xS product exhibits a hexagonal disk shape, some of which exhibit a face-to-face assembly behavior.23,24 The average size and thickness of the Cu1.2S nanodisks are measured to be 10.3 ± 1.8 nm and 5.9 ± 0.6 nm by statistically evaluating 100 particles, respectively, without any size sorting or post-preparation fractionation (Fig. 1a). Upon further decreasing the injection volumes to 17 and 14 mL, no obvious change is observed in the morphology (Fig. 1b and c). When the injection volume decreases to 12 mL, the hexagonal shape disappears and the as-prepared Cu1.94S NCs are mainly in a spherical morphology and the average diameter is 7.7 ± 0.5 nm (Fig. 1d). The corresponding HRTEM images of Cu2−xS NCs are shown in Fig. 1. Due to the rearrangement of Cu and S atoms, phase changes may result in dimensional and morphological changes. Different crystal facets lead to the change in surface energy, affecting the nucleation and growth speed.25,26 | ||
| Fig. 1 TEM images of Cu2−xS nanostructures: (a) Cu1.2S, (b) Cu1.4S, (c) Cu1.75S, (d) Cu1.94S NCs. Insets: Higher magnification of individual Cu2−xS NCs and the scale bar in the insets corresponds to 5 nm. | ||
The XRD technique was used to study the crystal structure of Cu1.2S, Cu1.4S, Cu1.75S, and Cu1.94S NCs, as shown in Fig. 2a. When injecting 21 mL of the S–OA solution into the Cu–OM solution, four strong characteristic diffraction peaks are observed in the XRD patterns, which can be indexed to the (1 0 2), (1 0 3), (0 0 6) and (1 1 0) planes of covellite (JCPDS no. 06-0464), confirming the formation of a hexagonal Cu1.2S phase. As the injection volume decreased to 17 mL, there is a strong diffraction peak located at 2θ = 38° and the broadening of the diffraction peak appears in the range of 2θ = 46–50° (Fig. 2b), which suggests that the crystal phase transforms to roxbyite. Further decreasing the injection volume of the S–OA solution to 14 mL, a relatively strong diffraction peak at 2θ = 46.71° and a relatively weak peak at 2θ = 48.71° are observed in the XRD patterns (Fig. 2b), which are assigned to the (0 16 0) and (8 8 6) planes of the roxbyite Cu7S4 phase (JCPDS no. 23-0958). This reveals that the crystal phase transforms from the covellite to the roxbyite phase with the decrease of S. Compared to the XRD peaks of Cu1.75S NCs, the intensity of the diffraction peak at 2θ = 48.71° of Cu1.94S NCs becomes stronger. This peak together with other diffraction peaks match well with that of Cu31S16 of the djurleite phase (JCPDS no. 23-0959). In this way, we can easily manipulate the doping level of the Cu2−xS NCs by varying the amount of the S–OA precursor solution.
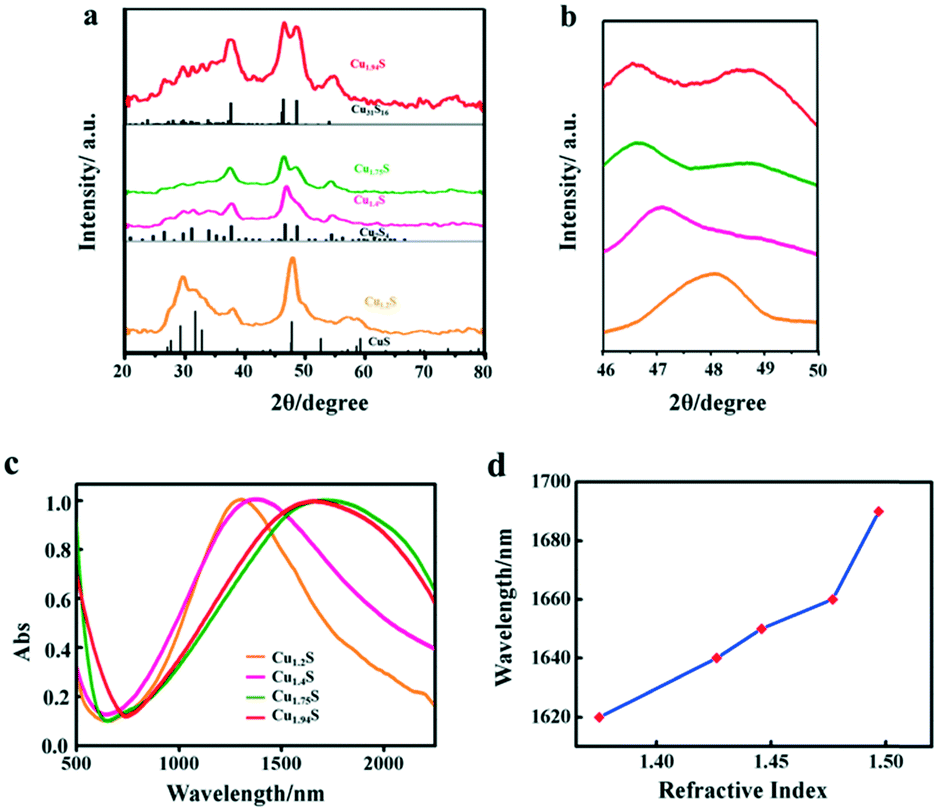 | ||
| Fig. 2 (a) XRD patterns of a series of Cu2−xS nanostructures and the bottom vertical lines are the standard diffraction peaks of hexagonal CuS (covellite, JCPDS no. 06-0464), monoclinic Cu7S4 (roxbyite, JCPDS no. 23-0958) and monoclinic Cu31S16 (djurleite, JCPDS no. 23-0959). (b) The corresponding XRD patterns in the 2θ angle range from 46° to 50°. (c) NIR LSPR absorption spectra of four Cu2−xS nanostructures (in TCE). (d) Dependence of the LSPR absorbance peak wavelength on the solvent refractive index for Cu1.94S NCs. | ||
The free holes in the p-doped semiconductor NC generate a strong LSPR. The LSPR energy, which is influenced by the copper content, directly depends on the free carrier concentration. The LSPR in Cu2−xS was measured by UV–vis–NIR absorption spectroscopy. Fig. 2c shows the LSPR absorption spectra of four kinds of Cu2−xS products, dispersed in TCE. For the covellite Cu1.2S NCs, a well-defined NIR absorption peak at 1270 nm is observed in Fig. 2c. While with the decrease of S percentage to form Cu1.4S NCs, the peak shows a red shift from 1270 to 1350 nm. The red shift is mainly because the crystal phase change from covellite to roxbyite. With the further decreased injection volume to form Cu1.75S and Cu1.94S NCs, there is a blue shift of the LSPR wavelength from 1690 nm to 1660 nm. This is caused by a combined influence of phase transformation and copper vacancy which together caused the variation of the carrier concentration. Here, we found that the LSPR in these Cu2−xS NCs is tunable by simply changing the injection amount of the S–OA solution during the synthesis of copper sulfide NCs. To confirm that the observed absorbance came from LSPR, we characterized its dependence on the refractive index of the surroundings environment of the NCs. The NIR absorbance gradually red shifts with increasing refractive index of the solvent in which Cu1.94S was dispersed (Fig. 5d). The peak of the LSPR absorbance red shifted 70 nm from R = 1.375 (hexane) to R = 1.4969 (toluene). As the refractive index of the solvent increases, this red-shift in the NIR absorbance is the hallmark behavior of LSPR. The resonance in polarizability of the NC is associated with the surrounding environment and the relative dielectric constants of the NC.27,28
In order to further clarify the composition and valence state of Cu and S in the obtained products of different Cu2−xS NCs, XPS analysis was performed. Fig. 3 shows the high-resolution XPS spectra of Cu 2p and S 2p of the products synthesized by using S–OA solution volumes of 21 and 12 mL. Cu 2p1/2 and Cu 2p3/2 corresponding to two binding energy peaks at 952.4 and 932.6 eV are observed in the Cu 2p spectra, respectively. It is worth noting that, as the injection volume of the S–OA solution decreases from 21 to 12 mL, the intensity of the satellite peak becomes weaker. Compared to the Cu 2p spectra, the S 2p spectra undergo an obvious change from Cu1.2S to Cu1.94S (Fig. 3b). There are disulfide bonds in covellite CuS to balance the charge and maintain the stability in the unit cell.26,29 As shown in the deconvolution of the S element in Fig. S1,† there are two main doublets, sulfide (S 2p3/2 at a BE of 161.5 eV and S 2p1/2 at a BE of 162.7 eV) and disulfide (S 2p3/2 at a BE of 162.3 eV and S 2p1/2 at a BE of 163.5 eV) in Cu1.2S. For each doublet, a spin–orbit splitting of 1.2 eV and an S 2p3/2 S 2p1/2 branching ratio of 2 were used. The ratio of the disulfide area and sulfide area was 2.4:1, approximately consistent with pure covellite. The disulphide content decreased in Cu1.94S sharply, which should be zero theoretically. The peak moved from 162.3 and 163.5 eV (sulphide) to 161.5 and 162.7 eV (disulphide) separately, indicating that the Cu–S bond increases from the covellite to djurleite phase.30
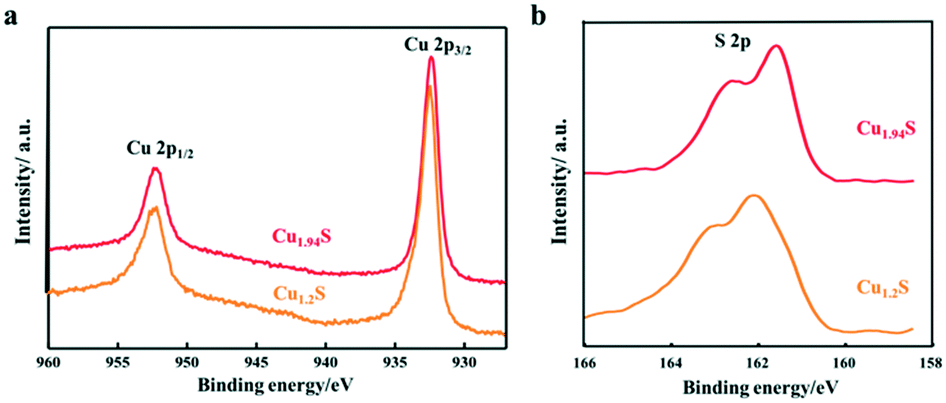 | ||
| Fig. 3 XPS spectra of the Cu2−xS NCs synthesized by using different volumes of the S–OA solution: (a) Cu 2p and (b) S 2p. | ||
Photocatalytic activities of Cu2−xS NCs
Many studies have reported that Cu2−xS microstructures with different morphologies have been used as photocatalysts to degrade organic pollutants in water.1,18–20,31 As we know, the photodegradation of dyes depends on the adsorption of dyes over the catalyst, and the larger the surface area of the catalyst, the higher the adsorption. There is no doubt that the specific surface area depends on the size of the NCs i.e. the smaller the size, the larger the specific surface area. With these facts in mind, these four kinds of photocatalysts are designed with gradually decreased sizes, and their photocatalytic activity in decomposing different organic dyes are monitored. The characteristic absorption peaks of organic dye molecules were used to monitor the degradation of dyes with the extension of irradiation time. The absorbance peak intensity gradually decreases at the corresponding wavelength of dyes with the irradiation time, indicating the degradation of dye molecules.In order to verify the photocatalytic properties, four different vacancy-doped NCs (Cu1.2S, Cu1.4S Cu1.75S and Cu1.94S NCs) were used as photocatalysts to degrade two kinds of organic dyes. Fig. 4a shows the UV–vis absorption spectra of a series of RhB solutions after being treated with Cu1.2S NCs with different irradiation times. In the case of RhB, which is a cationic dye and possesses an active positive nitrogen center, the intense peak at 554 nm is used to observe the overall degradation efficiency. It can be clearly seen that the intensity of the absorption peaks of RhB molecules weaken gradually and disappeared with extending the illumination time and 57% of the dye molecules were degraded with the exposure time of 120 min under UV light. Compared to Cu1.2S NCs, almost 80% degradation of RhB was achieved by Cu1.94S NCs in the first 20 min and continued degradation later, because initially quite a lot of active coordination sites are available for adsorption of dye molecules. Meanwhile after 120 min, the number of sites decreased. Finally, overall 95% of dyes were degraded (Fig. 4b). In the case of MO, which is an anionic dye, the intense peak at 455 nm was chosen to monitor the photocatalytic activity. The photocatalytic degradation efficiency of Cu1.94S NCs has reached 60% in the first 20 min though the rate slows down after (Fig. 4d). However, only 15% of dye degradation was observed in the presence of Cu1.2S NCs within the irradiation time of 120 min (Fig. 4c).
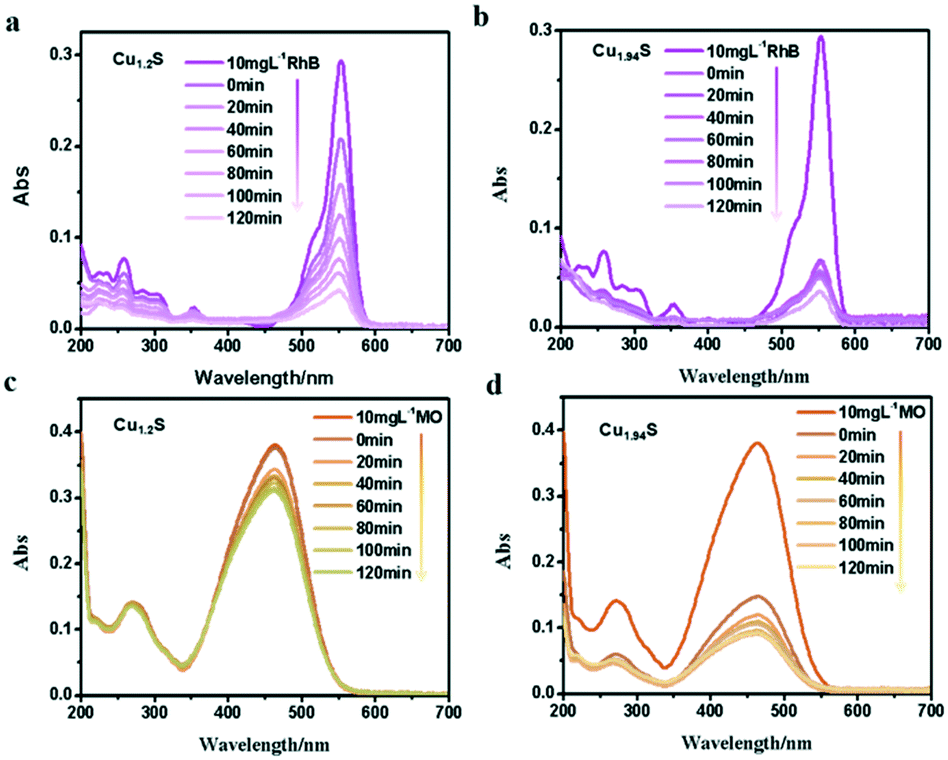 | ||
| Fig. 4 UV–visible spectra change of (a and b) RhB, (c and d) MO with Cu1.2S and Cu1.94S NCs as photocatalysts under UV irradiation, respectively. | ||
From the results above, we found that Cu1.94S NCs have a stronger adsorption and degradation capacity of dyes compared to Cu1.2S NCs. This indicates that the nature of the active site has changed due to an increase in copper deficiency. Since the morphology and size of the two samples were almost identical, the catalytic activity measured with Cu1.94S NCs was greatly increased due to their significantly higher free hole concentration. In addition, the photocatalytic degradation efficiency for RhB solutions is better than that for the MO dye. Beyond that, the UV visible absorption of dyes degraded with other Cu2−xS NCs is shown in Fig. S2.† We can see that Cu1.4S and Cu1.75S NCs also have more favorable photocatalytic activities for degrading RhB compared to those for the MO dye. According to the comprehensive experimental results, it can be concluded that: 1. the degradation process is quite fast for cationic dyes, like RhB, but quite slow for anionic dyes, like MO. 2. The order of NC adsorption capacity is about Cu1.94S ≈ Cu1.75S ≫ Cu1.4S ≈ Cu1.2S, whose reason will be elucidated in detail later.
The photocatalytic degradation rate of RhB and MO over four photocatalysts is shown in Fig. 5a and b. For example, the degradation efficiency of RhB in the cases of Cu1.94S and Cu1.75S achieves 75% after an irradiation time of 20 min, but Cu1.4S and Cu1.2S only reach about 40%, suggesting that the former materials have more excellent photocatalytic efficiency than the latter NCs for the degradation of RhB, and this efficiency is related to their surface areas. The same experimental result appears in MO (in the cases of Cu1.94S and Cu1.75S achieving 65% and 50%, respectively, and Cu1.4S and Cu1.2S only reach about 10% after an irradiation time of 20 min). It is generally believed that the photocatalytic process is mainly related to the adsorption and desorption of the catalyst surface. A high specific surface area in the case of nano-photocatalysts is of principal concern for effective degradation, which results in more unsaturated surface coordination sites exposed to the reactants. The order of sizes is Cu1.94S < Cu1.75S < Cu1.4S < Cu1.2S, with the opposite specific surface area order, leading to the adsorption capacity of dyes as Cu1.94S > Cu1.75S > Cu1.4S ≈ Cu1.2S.
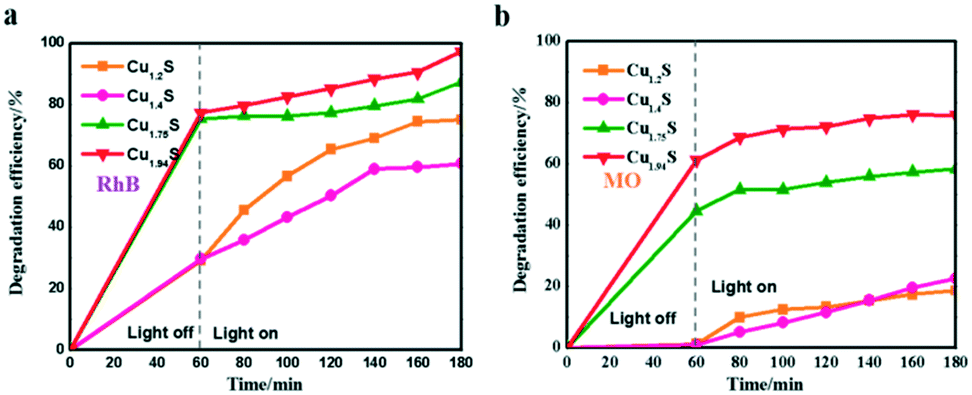 | ||
| Fig. 5 Photocatalytic activities of Cu2−xS for RhB (a) and MO (b) photodegradation under UV irradiation. | ||
It should be noted that when RhB was substituted by MO (Fig. 5a and 4b), Cu2−xS NCs showed quite poor catalytic activities, resulting in a low degradation of MO. For instance, the photocatalytic efficiencies of RhB and MO in the case of Cu1.2S are 70% and 18%, respectively, after being irradiated for 120 min. This may be explained by the fact that RhB is a kind of cationic dye,21,32–34 but MO is an anionic dye,14,35,36 and the former exhibits a higher affinity for the surface of Cu2−xS with negative charges. Therefore, the Cu2−xS NC catalysts can bring about a significant selective degradation of these two kinds of dyes. Herein, we will attempt to give a possible interpretation of the photocatalytic degradation of dyes in the presence of the Cu2−xS NCs. It is demonstrated by other reports that the surface of Cu2−xS has negative charges. As shown in Fig. 5a, the cationic dye, RhB, could initially be efficiently adsorbed onto the surface of the Cu2−xS NCs, especially, Cu1.94S NCs with a high specific surface area, in a dark environment after being fully mixed. The spontaneous adsorption process might be attributed to an ion–dipole interaction between the localized negative charges of the sulfur ions (Sδ−) on the Cu2−xS surface and the N+ atoms with the RhB molecules.37,38 In addition, the electrostatic field in the negatively charged catalyst particles enables the storage of more cationic reactant molecules. The surface possibly has a negative charge, OH−, the transformation of surface bound oxygen that is adsorbed during the process.11,12 In our work, zeta potentials of Cu2−xS NCs in water solution were recorded at pH 7.0 as shown in Fig. S3.† Cu1.2S, Cu1.4S, Cu1.75S and Cu1.94S NCs bear a negative charge, with values of about −22, −20, −21.1 and −11.4 mV, respectively. We can deduce that the monodispersity of Cu2−xS NCs at this stage is the best. Therefore, the enhancement of the catalytic activity by a high surface area is reasonable. In this way, the anionic surface of the catalyst attracts the cationic dye molecule by the electrostatic field force and electron transfer after irradiation is facilitated. In contrast, MO, an anionic dye, could not efficiently be adsorbed onto the surface of the catalyst, resulting in a very low degradation rate. However, the dye molecules get aggregated and become slightly polarizable and hence electron transfer becomes a possibility to a certain extent.
Furthermore, it can be seen that the order of the photocatalytic activity for the degradation of RhB is about Cu1.94S > Cu1.75S > Cu1.2S > Cu1.4S, and the order of the photocatalytic performance for the degradation of MO is about Cu1.94S > Cu1.75S ≫ Cu1.4S ≈ Cu1.2S. In general, photocatalytic materials with large free carrier concentrations lead to high photocatalytic activity. Three factors are closely associated with it: (1) the carrier concentration is already high without photoexcitation because of the existence of Cu vacancies in the Cu2−xS lattice. The different doping levels of Cu contributes to the LSPR energy, which is directly affected by the free carrier concentration. The doping levels of Cu increases with the copper vacancy increasing from Cu1.94S to Cu1.2S.8,39 (2) The Cu vacancies may tend to trap photoexcited delocalized electrons. The Cu deficiencies may also become a recombination center of electrons and holes, and as the doping levels of Cu increases from Cu1.94S to Cu1.2S, the free carrier concentration decreases.8,39 (3) The injection volume of the S–OA solution also has an effect on the LSPR energy on account of the negative charge carried by deprotonated oleic acid. Coordination of deprotonated carboxyl groups to the surface may be caught in free holes, leading to the decrease of the effective free carrier concentration. As the injection amount of the S–OA solution decreases from 17 mL to 12 mL during synthesis, the LSPR energy gradually increases, leading to a higher photocatalytic activity from Cu1.2S to Cu1.94S. However, the uncharged oleylamine ligands do not have such a remarkable effect on the concentration of free carriers (Fig. 6).40 (4) In addition, we can see that the adsorption capacity of Cu1.94S and Cu1.75S NCs is significantly stronger than Cu1.4S and Cu1.2S, either for cationic dyes or anionic dyes. According to previous studies, different doping levels change the nature of the active sites, so we suspect that the heavily doped Cu2−xS NCs have poor adsorption capacity, which also indirectly affecting the photocatalytic performance of photocatalysts.41 Photocatalytic activity mainly depends on the combined effect of the four factors above.
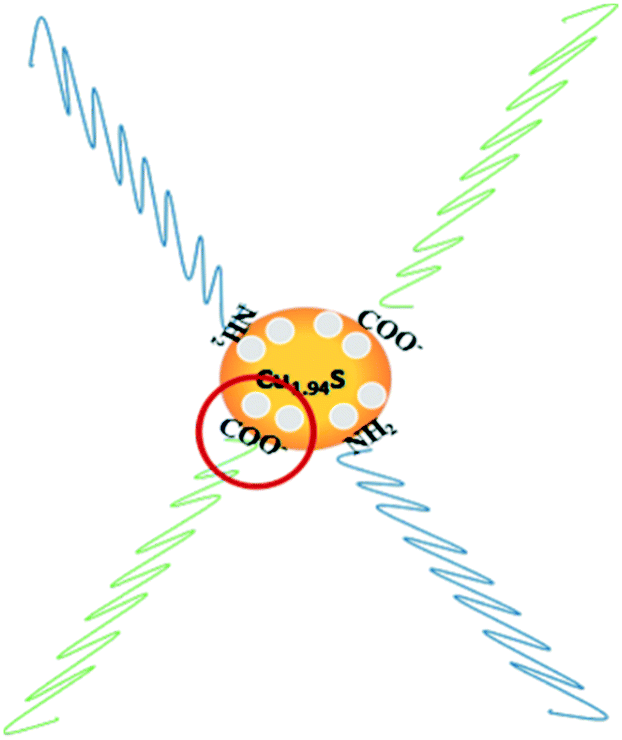 | ||
| Fig. 6 Schematic illustration for the mechanism of carrier density reduction by coordination of deprotonated carboxyl groups to the NC surface. | ||
Thus, it is necessary to theoretically calculate the specific free carrier concentration, closely related with LSPR absorption, which can be described by the Mie–Drude model.42–44 Herein the carrier density was calculated with this model, as shown in the detailed discussion in the ESI. According to the results, the data of free carrier density are summarized in Table 1, which are in accordance with the experimental phenomenon. According to above experimental results and theoretical calculations, it can be confirmed that the photocatalytic activity could be regulated and controlled by varying the free carrier concentration of Cu2−xS NCs.
| Cu2−xS | Cu1.2S | Cu1.4S | Cu1.75S | Cu1.94S |
|---|---|---|---|---|
| ω sp (eV) | 1.00 | 0.90 | 0.81 | 0.86 |
| ω p (eV) | 2.12 | 2.06 | 2.31 | 2.44 |
| N h (1021 cm−3) | 2.64 | 2.50 | 3.14 | 3.50 |
Degradation kinetic studies
Based on the Langmuir Hinshelwood (L–H) equations,45,46 a kinetic model with Cu1.2S, Cu1.4S, Cu1.75S and Cu1.94S NCs as photocatalysts could be used appropriately to present the values of the reaction rate for the dye degradation in dilute solutions. The kinetic curves for degrading dyes with four photocatalysts under ultraviolet light is expressed with the function, −ln(C/C0) = kt, where C and C0 are the instantaneous and initial concentrations of the dye solution, respectively and k is the apparent reaction rate constant (min−1). Fig. 7a and b show that all the catalytic reactions fit well with the pseudo-first order correlation. Consistent with the photocatalytic activity curves, it is observed that the kinetics is faster for the cationic dye molecules (RhB) than anionic dyes (MO).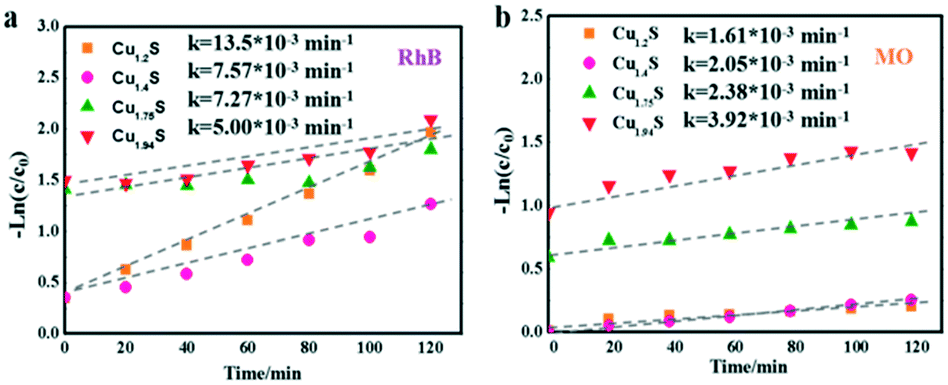 | ||
| Fig. 7 Kinetic curves of the Cu2−xS NCs for RhB (a) and MO (b) photodegradation under UV irradiation. | ||
Mechanism of degradation
Scheme 1 shows the mechanism of the photocatalytic reaction with Cu2−xS NCs. Electron–hole pairs are formed by electrons from the valence band which are excited to the conduction band under ultraviolet radiation. The as-obtained holes as well as intrinsic holes caused by Cu vacancies exhibit exceptional oxidizing properties. Some free electrons are transferred to the catalyst surface reducing O2 to strong oxidizing O2−. Meanwhile the holes oxidize H2O into hydroxyl radicals. These radicals exhibit intense oxidizing properties to destroy organic pollutant molecules.47 In this process, electrons and holes, known collectively as free carriers, play a key role. Varying the x value brings different intrinsic LSPR energies, i.e. holes, increasing the oxidizing property to pollutants, but meanwhile these deficiencies turn into recombination centers of electrons and holes. And deprotonated carboxylic functional groups of OA quench the excess free holes on the surface of Cu2−xS NCs to decrease the LSPR energy, which significantly effects the free carrier concentration. In addition, the nature of the active site has also changed due to an increase in copper deficiency. Consequently, Cu2−xS NCs with different doping levels do have diverse photocatalytic activities.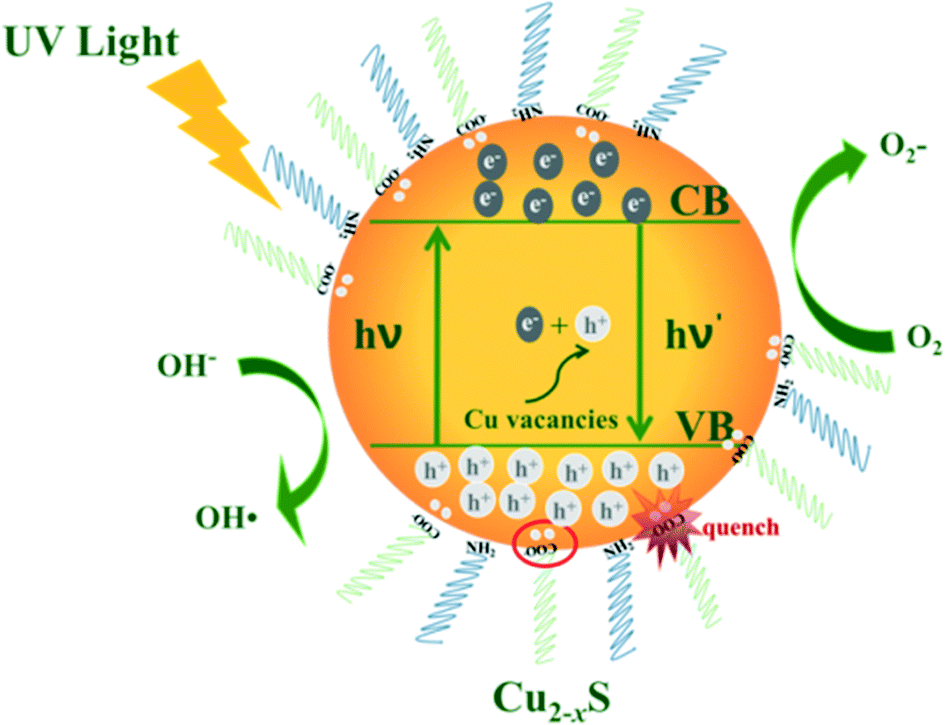 | ||
| Scheme 1 Mechanism of photocatalytic reaction. | ||
In addition, the stability of photocatalysts is a critical practical issue. After the first cycle of the photodegradation, we failed to collect even half of the catalysts from the dye solution mainly because of their small size. Hence, it was difficult to conduct another photocatalytic experiment. However, we verified the stability in terms of the morphology and optical behaviour of the used photocatalyst. The (HR)TEM and NIR absorption spectra of the Cu1.94S nanocatalyst collected after the first photocatalyst cycle from the RhB solution are shown in Fig. S4.† The recycled Cu1.94S NCs show a NIR absorption peak at 1620 nm, which barely moved compared to the original Cu1.94S photocatalyst. The morphology and size of the Cu1.94S NCs did not change after a photocatalytic process (Fig. S4b†). The TEM image of Cu1.94S NCs show that the nanocatalyst is still highly crystalline and directional. There is an obvious interplanar distance of 0.19 nm in the HRTEM images of the Cu1.94S NCs, corresponding to the (10 5 2) plane of djurleite. According to the morphology and optical test, the as-prepared Cu1.94S NCs exhibit good photocatalytic stability in the photodegradation of the RhB solution. It showed that the Cu2−xS NCs have potential application in wastewater treatment fields.
Conclusions
In summary, the injection amount of the S–OA precursor supplied was shown to control the free carrier concentration and LSPR energy by changing the copper content. The photocatalytic performance of Cu-deficient Cu2−xS NCs with tunable plasmonic properties was mainly investigated for the degradation of two kinds of dyes under ultraviolet light. It could be found that Cu1.94S NCs shows the best photocatalytic activity, followed by Cu1.75S, and then Cu1.4S and Cu1.2S, in two kinds of dyes. The carrier concentration upon the generation of Cu vacancies is the principal cause of that phenomenon. The experimental data show that the photocatalytic activity could be regulated and controlled by adjusting the carrier concentrations and could change the active sites nature of the photocatalysts. In addition, the Cu2−xS NC catalysts can bring about a significant selective degradation of these two kinds dyes. This research may open up a new avenue for the development of alternative non-precious metal catalysts by exploring new copper-deficient sulfides rich in free holes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (21908161) and the National Key R&D Program of China (2017YFB0404701).References
- Y. Liu, Y. Deng, Z. Sun, J. Wei, G. Zheng, A. M. Asiri, S. B. Khan, M. M. Rahman and D. Zhao, Small, 2013, 9, 2702–2708 CrossRef CAS PubMed.
- X. Bu, D. Zhou, J. Li, X. Zhang, K. Zhang, H. Zhang and B. Yang, Langmuir, 2014, 30, 1416–1423 CrossRef CAS PubMed.
- M. Zhou, R. Zhang, M. Huang, W. Lu, S. Song, M. P. Melancon, M. Tian, D. Liang and C. Li, J. Am. Chem. Soc., 2010, 132, 15351–15358 CrossRef CAS PubMed.
- Y. Wu, C. Wadia, W. Ma, B. Sadtler and A. P. Alivisatos, Nano Lett., 2008, 8, 2551–2555 CrossRef CAS PubMed.
- K. Vinokurov, O. Elimelech, O. Millo and U. Banin, ChemPhysChem, 2016, 17, 675–680 CrossRef CAS PubMed.
- S.-W. Hsu, W. Bryks and A. R. Tao, Chem. Mater., 2012, 24, 3765–3771 CrossRef CAS.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- I. Kriegel, J. Rodríguezfernández, A. Wisnet, H. Zhang, C. Waurisch, A. Eychmüller, A. Dubavik, A. O. Govorov and J. Feldmann, ACS Nano, 2013, 7, 4367–4377 CrossRef CAS PubMed.
- A. Wolf, T. Härtling, D. Hinrichs and D. Dorfs, ChemPhysChem, 2016, 17, 717–723 CrossRef CAS PubMed.
- D. Dorfs, T. Härtling, K. Miszta, N. C. Bigall, M. R. Kim, A. Genovese, A. Falqui, M. Povia and L. Manna, J. Am. Chem. Soc., 2011, 133, 11175–11180 CrossRef CAS PubMed.
- L. Shan, Y. Liu, J. Bi, J. Suriyaprakash and Z. Han, J. Alloys Compd., 2017, 721, 784–794 CrossRef CAS.
- W. Wang, D. Xu, B. Cheng, J. Yu and C. Jiang, J. Mater. Chem. A, 2017, 5, 5020–5029 RSC.
- J. Zhang, W. Li, Y. Li, L. Zhong and C. Xu, Appl. Catal., B, 2017, 217, 30–36 CrossRef CAS.
- E. Shin, S. Jin, J. Kim, S.-J. Chang, B.-H. Jun, K.-W. Park and J. Hong, Appl. Surf. Sci., 2016, 379, 33–38 CrossRef CAS.
- Y. Gu, C. Sun, C. Zhang, X. Luo, C. Xue and L. Zhao, CrystEngComm, 2019, 21, 5482–5491 RSC.
- B. Mulder, Phys. Status Solidi A, 1973, 18, 633–638 CrossRef CAS.
- X. Song and L. Gao, J. Phys. Chem. C, 2008, 112, 15299–15305 CrossRef CAS.
- J. Kundu and D. Pradhan, ACS Appl. Mater. Interfaces, 2014, 6, 1823–1834 CrossRef CAS PubMed.
- Z. Cheng, S. Wang, Q. Wang and B. Geng, CrystEngComm, 2010, 12, 144–149 RSC.
- Z. K. Yang, L. X. Song, Y. Teng and J. Xia, J. Mater. Chem. A, 2014, 2, 20004–20009 RSC.
- M. Basu, A. K. Sinha, M. Pradhan, S. Sarkar, Y. Negishi and T. Pal, Environ. Sci. Technol., 2010, 44, 6313–6318 CrossRef CAS PubMed.
- D. Zhu, A. Tang, L. Peng, Z. Liu, C. Yang and F. Teng, J. Mater. Chem. C, 2016, 4, 4880–4888 RSC.
- H. Wu and W. Chen, Nanoscale, 2011, 3, 5096–5102 RSC.
- Y. Liu, M. Liu and M. T. Swihart, Chem. Mater., 2017, 29, 4783–4791 CrossRef CAS.
- L. Liu, H. Zhong, Z. Bai, T. Zhang, W. Fu, L. Shi, H. Xie, L. Deng and B. Zou, Chem. Mater., 2013, 25, 4828–4834 CrossRef CAS.
- Y. Xie, L. Carbone, C. Nobile, V. Grillo, S. D'Agostino, S. F. Della, C. Giannini, D. Altamura, C. Oelsner and C. Kryschi, ACS Nano, 2013, 7, 7352–7369 CrossRef CAS PubMed.
- A. Comin and L. Manna, Chem. Soc. Rev., 2014, 45, 3957–3975 RSC.
- X. Liu and M. T. Swihart, Chem. Soc. Rev., 2014, 43, 3908–3920 RSC.
- Y. Liu, D. Yin and M. T. Swihart, Chem. Mater., 2018, 30, 8089–8098 CrossRef CAS.
- L. Zhao, X. Wang, F. Y. Fei, J. Wang, Z. Cheng, S. Dou, J. Wang and G. J. Snyder, J. Mater. Chem. A, 2015, 3, 9432–9437 RSC.
- Y. Hu, L. Zheng and Y. Sun, Catal. Today, 2014, 225, 177–184 CrossRef CAS.
- T. P. M. Chu, N. T. Nguyen, T. L. Vu, T. H. Dao, L. C. Dinh, H. L. Nguyen, T. H. Hoang, T. S. Le and T. D. Pham, Materials, 2019, 12, 450 CrossRef CAS PubMed.
- P. Qin, Y. Yang, X. Zhang, J. Niu, H. Yang, S. Tian, J. Zhu and M. Lu, Nanomaterials, 2018, 8, 1–14 Search PubMed.
- Y. Li, F. Liu, H. Zhang, X. Li, X. Dong, G. Lu and C. Wang, Mater. Lett., 2019, 238, 183–186 CrossRef CAS.
- T. S. Kazeem, M. Zubair, M. Daud, N. D. Mu'azu and M. A. Al-Harthi, Korean J. Chem. Eng., 2019, 36, 1057–1068 CrossRef CAS.
- Z. Wang, C.-Y. Zhu, H.-S. Zhao, S.-Y. Yin, S.-J. Wang, J.-H. Zhang, J.-J. Jiang, M. Pan and C.-Y. Su, J. Mater. Chem. A, 2019, 7, 4751–4758 RSC.
- Z. Liu, X. Liu, Y. Du, J. Ren and X. Qu, ACS Nano, 2015, 9, 10335–10346 CrossRef CAS PubMed.
- Y. Li, W. Lu, Q. Huang, C. Li and W. Chen, Nanomedicine, 2010, 5, 1161–1171 CrossRef CAS PubMed.
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef CAS PubMed.
- X. Liu, X. Wang, B. Zhou, W. C. Law, A. N. Cartwright and M. T. Swihart, Adv. Funct. Mater., 2013, 23, 1256–1264 CrossRef CAS.
- X. Wang, Y. Ke, H. Pan, K. Ma, Q. Xiao, D. Yin, G. Wu and M. T. Swihart, ACS Catal., 2015, 5, 2534–2540 CrossRef CAS.
- A. Venkadesh, S. Radhakrishnan and J. Mathiyarasu, Electrochim. Acta, 2017, 246, 544–552 CrossRef CAS.
- J. A. Faucheaux, A. L. Stanton and P. K. Jain, J. Phys. Chem. Lett., 2014, 5, 976–985 CrossRef CAS PubMed.
- D. Zhu, A. Tang, H. Ye, M. Wang, C. Yang and F. Teng, J. Mater. Chem. C, 2015, 3, 6686–6691 RSC.
- S. Šegota, L. Ćurković, D. Ljubas, V. Svetličić, I. F. Houra and N. Tomašić, Ceram. Int., 2011, 37, 1153–1160 CrossRef.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- M. Saranya, C. Santhosh, R. Ramachandran, P. Kollu, P. Saravanan, M. Vinoba, S. K. Jeong and A. N. Grace, Powder Technol., 2014, 252, 25–32 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce01501a |
| This journal is © The Royal Society of Chemistry 2020 |