
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystal structure features of CH3NH3PbI3−xBrx hybrid perovskites prepared by ball milling: a route to more stable materials†
Carlos Alberto
López
*ab,
María Consuelo
Alvarez-Galván
c,
María Victoria
Martínez-Huerta
c,
Francois
Fauth
 d and
José Antonio
Alonso
*a
d and
José Antonio
Alonso
*a
aInstituto de Ciencia de Materiales de Madrid, CSIC, Cantoblanco, 28049 Madrid, Spain. E-mail: ja.alonso@icmm.csic.es
bInstituto de Investigaciones en Tecnología Química (INTEQUI), UNSL, CONICET and Facultad de Química, Bioquímica y Farmacia, UNSL, Ej. De los Andes 950, San Luis, 5700, Argentine. E-mail: calclopez@gmail.com
cInstituto de Catálisis y Petroleoquímica, CSIC, Cantoblanco, 28049 Madrid, Spain
dCELLS–ALBA synchrotron, Cerdanyola del Valles, Barcelona, E-08290, Spain
First published on 25th November 2019
Abstract
Hybrid organic–inorganic perovskites, MAPbX3 (MA: methylammonium, CH3–NH3+; X = halogen), are active absorption materials in the new generation of solar cells. The triiodide specimen (MAPbI3) remains the most widely studied perovskite due to its ability to absorb broadband light below its band gap of 1.6 eV, but its degradation in a humid atmosphere has remained a major obstacle for commercialization. Here we found that CH3NH3PbI3−xBrx (x = 0.0, 1.0) perovskites, prepared by ball milling, exhibit superior stability, showing no signs of degradation after several months of exposure to humid air. A synchrotron X-ray diffraction (SXRD) investigation was useful to determine some peculiar structural features that may account for the improved stability. The crystal structure was analysed to be in the I4/mcm and acentric I4cm space groups, yielding similar agreement factors. In both, the inorganic framework presents a conspicuously lower tilting effect than that observed in samples prepared by wet methods; an additional difference arises since the tetragonal structure is stable down to 140 K. The orientation of the organic MA units may play an important role in the properties, given the degrees of freedom for internal motion of MA groups within the PbX6 network. From the refined C/N positions, the MA+ units lie along the c axis with 25% probability. The structure at 140 K shows that the MA+ contribution along the c axis vanishes at this temperature. By contrast, MAPbI2Br crystallizes in the cubic phase with the space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, also with a larger unit-cell volume than that previously described. The absence of phase transitions down to 120 K suggests that the anion disorder prevents the localization of MA units upon cooling.
m, also with a larger unit-cell volume than that previously described. The absence of phase transitions down to 120 K suggests that the anion disorder prevents the localization of MA units upon cooling.
Introduction
Organic–inorganic hybrid lead trihalide perovskites are highly promising semiconductors for both multi-junction photovoltaic and light emitting applications due to their tunable band gaps.1–3 Methylammonium lead triiodide perovskites (MAPbI3) have received much attention due to their ability to absorb broadband light below their band gap of 1.6 eV. However, MAPbI3 still faces several complex challenges that need to be addressed, where its thermal and moisture instability has remained a major obstacle for commercialization.4 In this regard, methods of tuning the halide anions of hybrid perovskites are the most common methods reported to seek a better system for photovoltaic applications, since they are also capable of band gap tuning by changing the composition of perovskites.5–9 The incorporation of bromine to produce a mixed halide hybrid perovskite (MAPbI3−xBrx) changes the physical properties, enhancing the stability of the material, but also increasing the band gap.5,8–10The morphology and the properties of mixed halide hybrid perovskites are highly sensitive to the adopted synthetic strategy and could be varied by carefully manipulating the solvent(s) and surface capping ligands. Several methods have been developed for producing hybrid perovskites including precipitation from solution and gas-phase deposition.1,11 In the solution-based method, stoichiometric mixtures of metal halides and organic ammonium halide precursors are normally dissolved in dimethylformamide (DMF) or dimethylsulfoxide (DMSO). In the gas-phase method, the precursors are generally co-evaporated in a vacuum deposition chamber onto a substrate by the use of two separate sources to form uniform and controlled-thickness films.
Mechanochemical synthesis has emerged as an attractive alternative solvent-free method, featuring remarkable simplicity, low cost, swiftness and reproducibility in line with exceptional green credentials (e.g. solvent-less solid-state synthesis) for the design of a wide range of perovskites.12–16 The combination of the mechanical energy generated under ball-milling conditions and the inherent chemical modification of structures/surfaces make this methodology extremely promising for alternative greener perovskite synthesis. Moreover, ball-milling procedures allow the formation of more stable perovskites, which are not degraded, forming MA4PbI6·2H2O or MA4PbI4Br2·2H2O, within a short time, by reaction with ambient humidity.16
Herein, two different polycrystalline hybrid perovskites, MAPbI3 and MAPbI2Br, have been successfully prepared by mechanochemical synthesis. The resultant polycrystalline powders were characterized by diffraction methods with synchrotron X-ray radiation, in complement with UV-vis spectroscopic measurements. Remarkably, these specimens were found to be stable in a laboratory air atmosphere for several months, which is ascribed to conspicuous structural features induced during the ball milling process.
Experimental
MAPbI3 and MAPbI2Br were obtained as microcrystalline powders from mechanochemical synthesis with stoichiometric amounts of PbI2 (or PbBr2) and MAI (or MABr). A total of 1.5 g of the reactants was milled using 30 zirconia balls of 5 mm diameter, with a final 8.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio, for 4 h at 400 rpm in a Retsch PM100 mill in an Ar atmosphere. For comparison purposes, MAPbI3 was also synthesized from a solution of stoichiometric amounts of PbI2 and MAI in dimethylformamide.16 Laboratory XRD patterns were collected on a Bruker D5 diffractometer with Cu Kα (λ = 1.5418 Å) radiation; the 2θ range was 4° up to 90° with increments of 0.03°. The absorption capacity of each perovskite powder was investigated by diffuse reflectance UV/vis spectroscopy. The UV-vis spectra of the materials were recorded with a UV-vis spectrometer equipped with a Varian Cary 3 UV-vis spectrometer equipped with an integration sphere. The synchrotron X-ray powder diffraction (SXRPD) patterns were collected in an MSPD high-resolution diffractometer at the ALBA facility, Barcelona (Spain), selecting an incident beam with a 38 keV energy, λ = 0.3252 Å. The high angular resolution mode (MAD set-up) on the MSPD-diffractometer was utilized.17 The polycrystalline powder was contained in quartz capillaries of 0.3 mm diameter, which were rotating during the acquisition time. SXRD patterns were collected at 140, 180, 298 and 393 K for MAPbI3 and at 120 and 298 K for MAPbI2Br. The refinement of the structure was performed by the Rietveld method using the Fullprof software.18,19 The following parameters were refined: zero-point error, scale factor, occupancy of the elements, atomic coordinates, and anisotropic displacements for all the atoms. The profile shape was modelled using the Thompson–Cox–Hastings pseudo-Voigt function20 and the instrumental resolution parameters were considered in the refinements in order to obtain the microstructural parameters. Thus, the u, v and w Caglioti parameters are fixed and correspond to the instrumental broadening, hence only the Lorentzian isotropic strain (X) and size (Y) parameters were refined. From these parameters, the apparent size and strain were calculated from the Scherrer and Stokes–Wilson formulae, respectively.
:1 mass ratio, for 4 h at 400 rpm in a Retsch PM100 mill in an Ar atmosphere. For comparison purposes, MAPbI3 was also synthesized from a solution of stoichiometric amounts of PbI2 and MAI in dimethylformamide.16 Laboratory XRD patterns were collected on a Bruker D5 diffractometer with Cu Kα (λ = 1.5418 Å) radiation; the 2θ range was 4° up to 90° with increments of 0.03°. The absorption capacity of each perovskite powder was investigated by diffuse reflectance UV/vis spectroscopy. The UV-vis spectra of the materials were recorded with a UV-vis spectrometer equipped with a Varian Cary 3 UV-vis spectrometer equipped with an integration sphere. The synchrotron X-ray powder diffraction (SXRPD) patterns were collected in an MSPD high-resolution diffractometer at the ALBA facility, Barcelona (Spain), selecting an incident beam with a 38 keV energy, λ = 0.3252 Å. The high angular resolution mode (MAD set-up) on the MSPD-diffractometer was utilized.17 The polycrystalline powder was contained in quartz capillaries of 0.3 mm diameter, which were rotating during the acquisition time. SXRD patterns were collected at 140, 180, 298 and 393 K for MAPbI3 and at 120 and 298 K for MAPbI2Br. The refinement of the structure was performed by the Rietveld method using the Fullprof software.18,19 The following parameters were refined: zero-point error, scale factor, occupancy of the elements, atomic coordinates, and anisotropic displacements for all the atoms. The profile shape was modelled using the Thompson–Cox–Hastings pseudo-Voigt function20 and the instrumental resolution parameters were considered in the refinements in order to obtain the microstructural parameters. Thus, the u, v and w Caglioti parameters are fixed and correspond to the instrumental broadening, hence only the Lorentzian isotropic strain (X) and size (Y) parameters were refined. From these parameters, the apparent size and strain were calculated from the Scherrer and Stokes–Wilson formulae, respectively.
The scanning electron microscope images were obtained on a Hitachi instrument, model TM-1000, coupled to an energy-dispersive X-ray (EDX) spectrometer, working with an acceleration voltage of 15 kV and 60 s of acquisition time. Chemical analysis was performed with a CHNS elemental analyzer PERKIN ELMER 2400.
Pb, I and Br were analysed by TXRF with a benchtop S2 PicoFox TXRF spectrometer from Bruker Nano (Germany), equipped with a molybdenum X-ray source working at 50 kV, 600 μA and 500 s (for quantitative analysis) with an XFlash SDD detector, effective area of 30 mm2 and an energy resolution better than 150 eV for Mn Kα. The Spectra 7 software package, also from Bruker, was used for control, acquisition, deconvolution, and integration of the signal peaks of all analyzed samples.
Results and discussion
Structural characterization
Chemical analysis (EDX, CHNS and TXRF) confirmed the expected stoichiometries (see the ESI†). The initial crystallographic identification was conducted using laboratory XRD. Fig. 1 shows the Le Bail fits of the XRD patterns. Both correspond to pure perovskites with a tetragonal symmetry for MAPbI3 and a cubic one for MAPbI2Br at RT.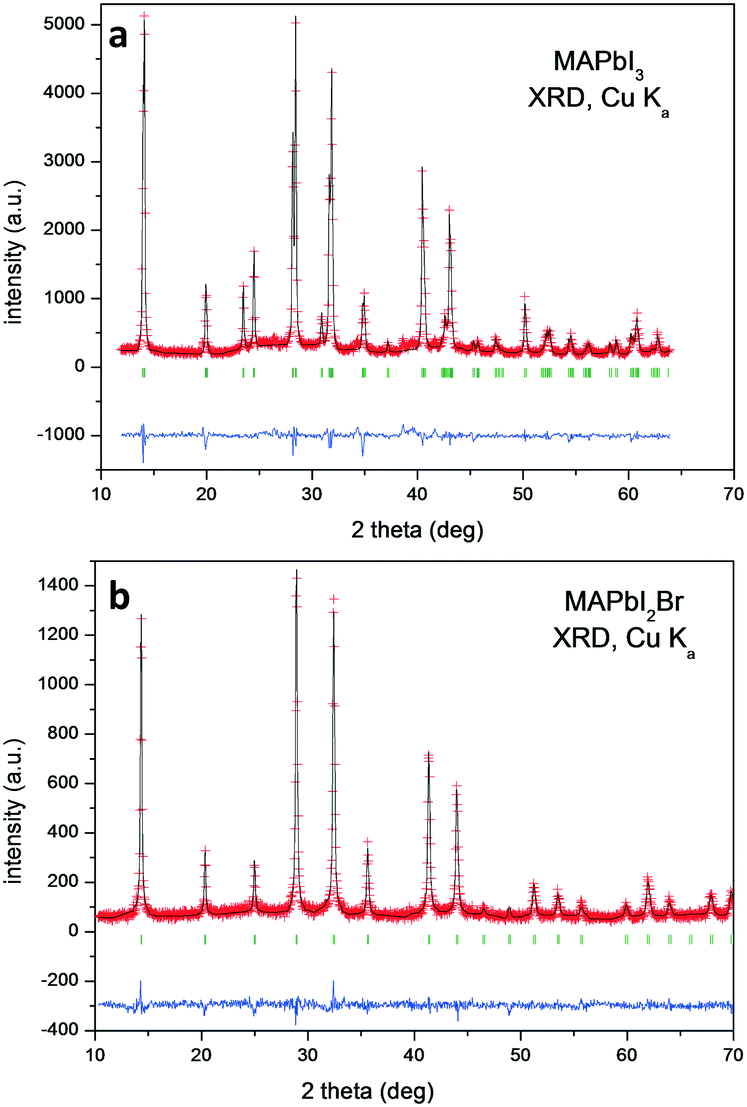 | ||
| Fig. 1 Le Bail refinement from laboratory XRD patterns at RT of a) MAPbI3 and b) MAPbI2Br. The red crosses, black lines, blue lines and green bars correspond to the observed intensity (Yobs), calculated intensity (Ycalc), difference (Yobs − Ycalc) and Bragg positions, respectively. | ||
MAPbI3
The tetragonal symmetry for MAPbI3 at RT is confirmed from synchrotron data; Fig. 2 shows the thermal evolution of the SXRD patterns. At 393 K, it becomes cubic, but between 140 K and 298 K, it remains tetragonal. However, there are some differences regarding the previously reported tetragonal space group.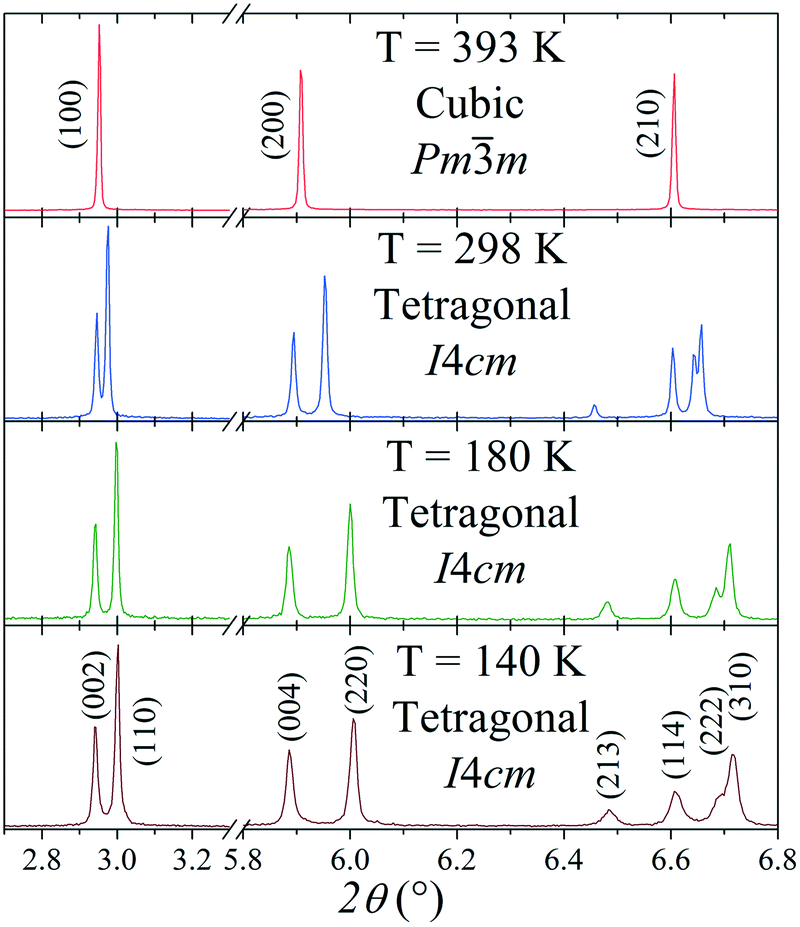 | ||
| Fig. 2 Thermal evolution of selected regions of the SXRD patterns of MAPbI3, in which a cubic–tetragonal phase transition is evidenced. | ||
The oldest structural report is authored by Poglitsch et al. in 1987 which described this phase in the I4/mcm space group at room temperature.21 Fifteen years later, Kawamura et al. confirmed this model from single crystal X-ray diffraction.22 Then, in 2013, two new studies analyzed the crystal structure: Baikie et al. confirmed this space group but they failed to model the location of the MA group.16 On the other hand, Stoumpos et al., also from single crystal X-ray diffraction, proposed the I4cm space group.23 In both cases, the unit-cell parameters are related to primitive cubic perovskite as 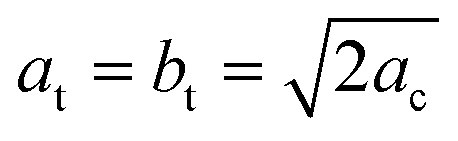 and ct = 2ac; however, the main difference lies in that I4/mcm is a centrosymmetric space group, in contrast to I4cm, with an off-center ferroelectric component along the c-axis. Then, in 2015, Weller et al., from neutron powder diffraction, reported this phase again in the I4/mcm space group.24 Regarding the MA+ orientation, Kawamura et al. found that the MA units are aligned along the [101]C, [
and ct = 2ac; however, the main difference lies in that I4/mcm is a centrosymmetric space group, in contrast to I4cm, with an off-center ferroelectric component along the c-axis. Then, in 2015, Weller et al., from neutron powder diffraction, reported this phase again in the I4/mcm space group.24 Regarding the MA+ orientation, Kawamura et al. found that the MA units are aligned along the [101]C, [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01]C, [0]C and [01]C directions of the cubic structure.22 This MA configuration is like the [110] orientation,25 but without the alignments on the (001) plane ([10]C and [110]C). Stoumpos et al., who proposed the I4cm space group, did not consider the MA+ disorder, refining a unique MA unit along the c-axis.23 Finally, the MA+ orientation reported by Weller et al.24 is similar to that reported by Kawamura et al.22
01]C, [0]C and [01]C directions of the cubic structure.22 This MA configuration is like the [110] orientation,25 but without the alignments on the (001) plane ([10]C and [110]C). Stoumpos et al., who proposed the I4cm space group, did not consider the MA+ disorder, refining a unique MA unit along the c-axis.23 Finally, the MA+ orientation reported by Weller et al.24 is similar to that reported by Kawamura et al.22
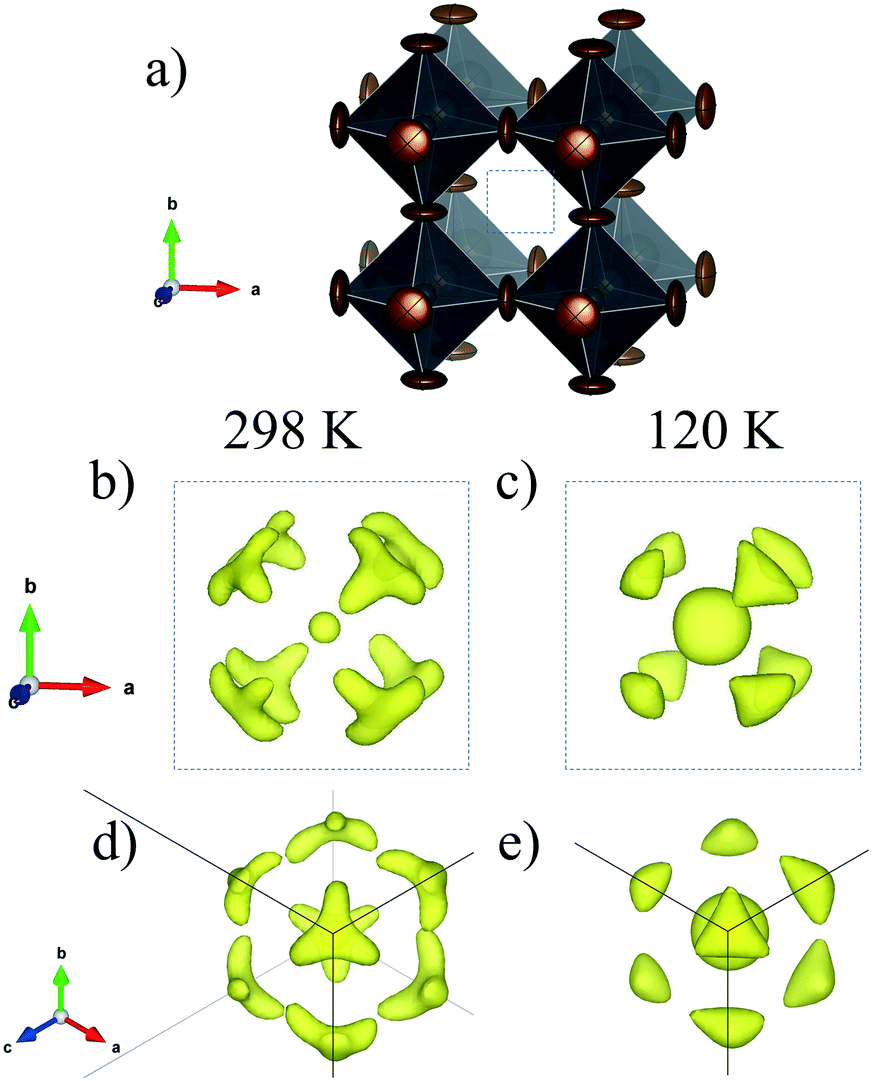
To analyze this dichotomy, we tested both models, I4/mcm and I4cm, on our SXRPD pattern at RT, placing first the lead and iodine atoms and then building difference Fourier maps (DFMs). The PbI6 lattice in the I4/mcm space group was assembled by placing Pb at the 4c (0, 0, 0) site and I at the 4a (0, 0, 1/4) and 8h (x, x + 1/2, 0) sites. For I4cm, Pb was placed in the 4a (0, 0, z) (with z fixed to 0) position and I was allocated in the 4a (0, 0, z) and 8c (x, x + 1/2, z) positions. As can be noted, the atomic coordinates are the same except for the z component, which is the polarizing direction in this non-centrosymmetric space group. At this point, the difference Fourier synthesis maps built for both symmetries reveal the missing electron density, which indicates that the MA units are clearly delocalized. Fig. 3 shows the PbI6 network in comparison with the electron density at 9e− for both space groups. From the shape and the intensity of these 3D plots, it is possible to infer the C/N positions. Six density maxima corresponding to C/N are observed, and these are well modeled with the following two sites for I4/mcm: (1, 1/2, z) and (x, x – 1/2, z). For I4cm, these sites are split into four: (1, 1/2, z), (1, 1/2, −z), (x, x – 1/2, z) and (−x, x – 1/2, −z). These observed positions were introduced, with the corresponding occupancy, and refined for both space groups. Both centrosymmetric and non-centrosymmetric space groups yielded good pattern fits, but the reliability factors are lower in the first case (I4/mcm: RBragg = 4.67%; I4cm: RBragg = 4.97%). Hence, the I4/mcm space group was selected as the more adequate model to describe MAPbI3 at RT. This is in agreement with recent studies from second harmonic generation (SHG),26,27 revealing that there is no evidence of non-centrosymmetric symmetry, thus confirming the I4/mcm space group in MAPbI3.
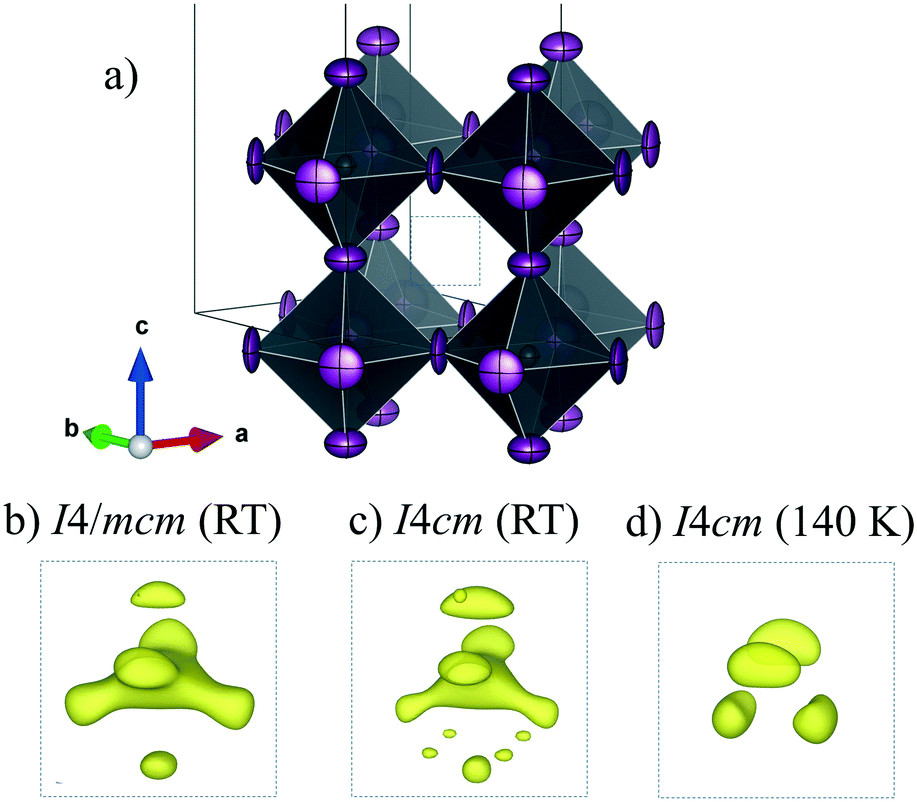 | ||
| Fig. 3 Schematic view of the PbI6 framework (a) and electron density for the I4/mcm (b) and I4cm (c) space groups at RT and I4/mcm at 140 K (d). | ||
The Rietveld plot from the SXRD pattern at RT is plotted in Fig. 4a, and the results of the crystallographic refinements are listed in Table 1. A schematic view of the tetragonal crystal structure is shown in Fig. 4b. From the refined C/N positions, it is possible to assume the possible alignments of MA+ units with respect to the c axis; hence, MA could be parallel or perpendicular (above or below the (1, 1/2, 1/4) position) as is highlighted in Fig. 4b. Considering the occupancy factors, it is possible to infer that MA+ is along the c axis with 25% probability. In order to study the crystal structure at lower temperatures, synchrotron patterns were collected at 180 K and 140 K and the same analytical procedures were performed.
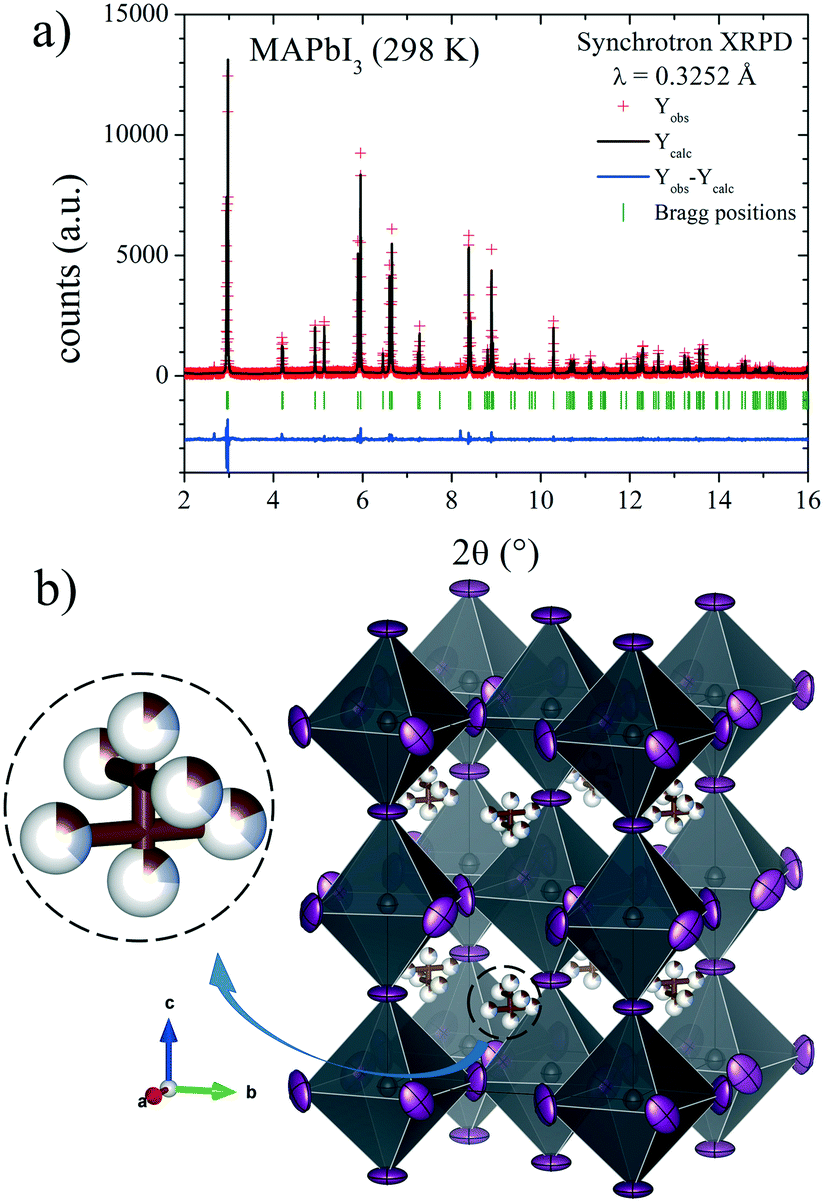 | ||
| Fig. 4 a) SXRD profile after Rietveld refinement and b) schematic view of MAPbI3 at RT in the I4/mcm space group. Only the C/N (undistinguishable) atoms of the MA units are shown. | ||
| x | y | z | U iso*/Ueq | Occ. | |
|---|---|---|---|---|---|
| Pb1 | 0 | 0 | 0 | 0.031(2) | 1 |
| I1 | 0 | 0 | 0.25 | 0.081(3) | 1 |
| I2 | 0.2854(3) | 0.7854(3) | 0 | 0.082(3) | 1 |
| C/N1 | 1 | 0.5 | 0.202(9) | 0.013* | 0.13(3)/0.13(3) |
| C/N2 | 0.933(5) | 0.433(5) | 0.231(5) | 0.013* | 0.18(2)/0.18(2) |
| Atomic displacement parameters (Å2) | ||||||
|---|---|---|---|---|---|---|
| U 11 | U 22 | U 33 | U 12 | U 13 | U 23 | |
| R p = 10.2%, Rwp = 13.4%, χ2 = 1.28, RBragg = 4.67%. a The numbers with the asterisk correspond to Uiso*, and the numbers without asterisk correspond to Ueq. | ||||||
| Pb1 | 0.031(1) | 0.031(1) | 0.030(2) | 0 | 0 | 0 |
| I1 | 0.109(3) | 0.109(3) | 0.024(3) | 0 | 0 | 0 |
| I2 | 0.065(2) | 0.065(2) | 0.117(4) | 0.042(3) | 0 | 0 |
The structures at 180 and 140 K can also be defined to be in the I4/mcm space group, although some distinct features concerning the orientation of the MA+ units were identified. The DFMs at 140 K (Fig. 3d) only reveal the electron density compatible with the (x, x – 1/2, z) and (−x, x – 1/2, −z) sites, showing that the MA+ contribution along the c axis vanishes at this temperature. The Rietveld refinements of the C/N positions and occupancy factors confirm the nonexistence of the MA+ orientation along the c axis at 140 and 180 K. Fig. 5a and b show the Rietveld refinement and a schematic view of the crystal structure at 140 K, respectively. Table S1 (ESI†) lists the main crystal data after the refinements at 180 and 140 K. The obtained C/N sites at 140 and 180 K show that the MA+ units can be found either above or below the (1, 1/2, 1/4) position. Besides, it is possible to observe that this MA+ displacement is concomitant with the PbI6 octahedral tilt. This fact is deduced from the analysis of the C/N–I distances. Fig. 5c and d show three non-equivalent distances: C–I1, C–I2 and C–I2′ with the following values: 3.89(3), 4.07(4) and 3.92(5) Å, respectively. These values can be considered equal within the experimental errors. A similar situation is found at 180 K (<C–I1>: 3.99(3) Å, <C–I2>: 4.14(4) Å and <C–I2′>: 4.03(5) Å). Despite ignoring the H positions and the H-bond interactions, it is possible to observe a reduction in the degree of freedom of MA+ units with respect to the RT structure. The tilting angles of the PbI6 octahedral framework also evidence this reduction; these values (estimated as φ = (180° − θ)/2, where θ = <Pb–I–Pb>) are 8.06°, 11.18° and 11.50° at 298, 180 and 140 K, respectively. There is an increase in the tilting angle as temperature decreases; however, this is not a linear increase as is usually observed for purely inorganic perovskites, supporting the reduction in the degree of freedom of MA+ at lower temperatures. Additionally, the tilt at RT is lower than that reported previously by Kawamura et al. (in I4/mcm)22 and Stoumpos et al. (in I4cm)23 whose tilts are 9.79° and 8.21°, respectively. In both cases, MAPbI3 was synthesized from wet methods. This difference allows inferring that the milling method yields a crystal structure closer to the ideal cubic symmetry, or in other words, with a tolerance factor28 closer to 1. This also would explain the fact that down to 140 K, the structure still remains tetragonal, in contrast to the reported phase transition temperatures to orthorhombic symmetry, at around 165 K.21,24 These behaviours can be related to an enhancement in the crystal structure stability.
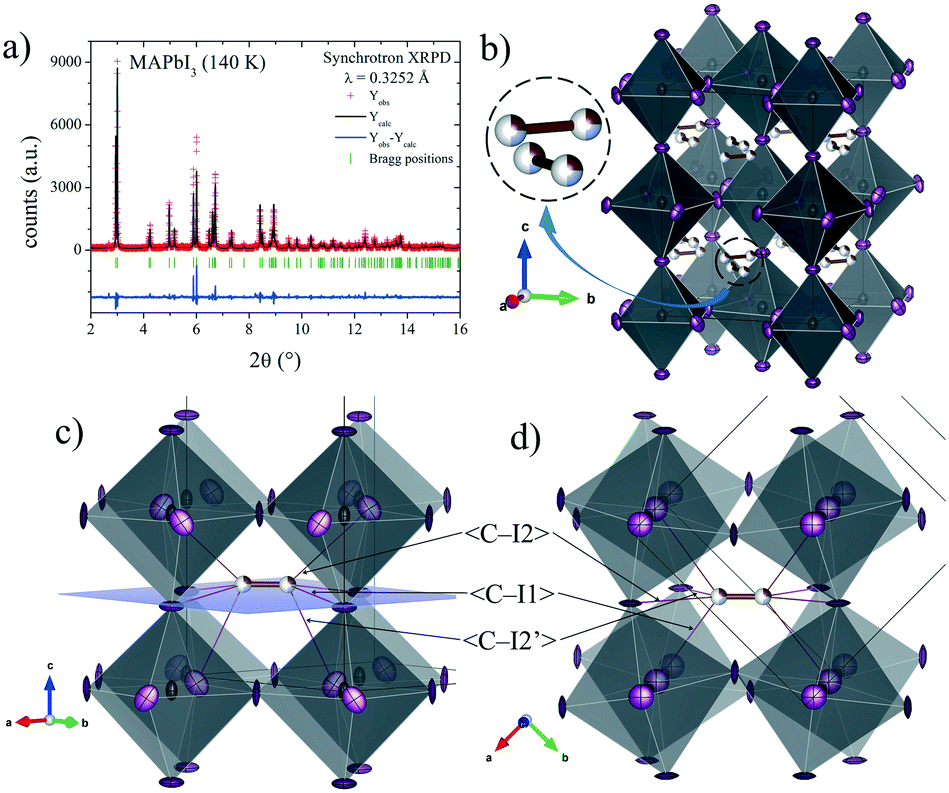 | ||
| Fig. 5 a) SXRD profile after Rietveld refinement and b) schematic view of MAPbI3 at 140 K in the I4/mcm space group. c) and d) Detailed views of main C/N–I distances of a MA unit in the PbI6 framework. | ||
Finally, the cubic symmetry defined in the space group Pmm, observed above room temperature (393 K) was also previously reported by Weller et al.24 They reported a phase transition to the cubic system at 327 K, describing a crystal structure at 352 K where the MA molecule was aligned along the (100) direction. However, our DFM at 393 K shows that the MA units are clearly aligned along the (110) direction; therefore, the C/N atoms are placed and could be refined at the Wyckoff site 12j (1/2, y, y). Fig. 6 shows the Rietveld refinement profile and views of the cubic crystal structure at 393 K. Table S2† lists the main crystallographic data at this temperature.
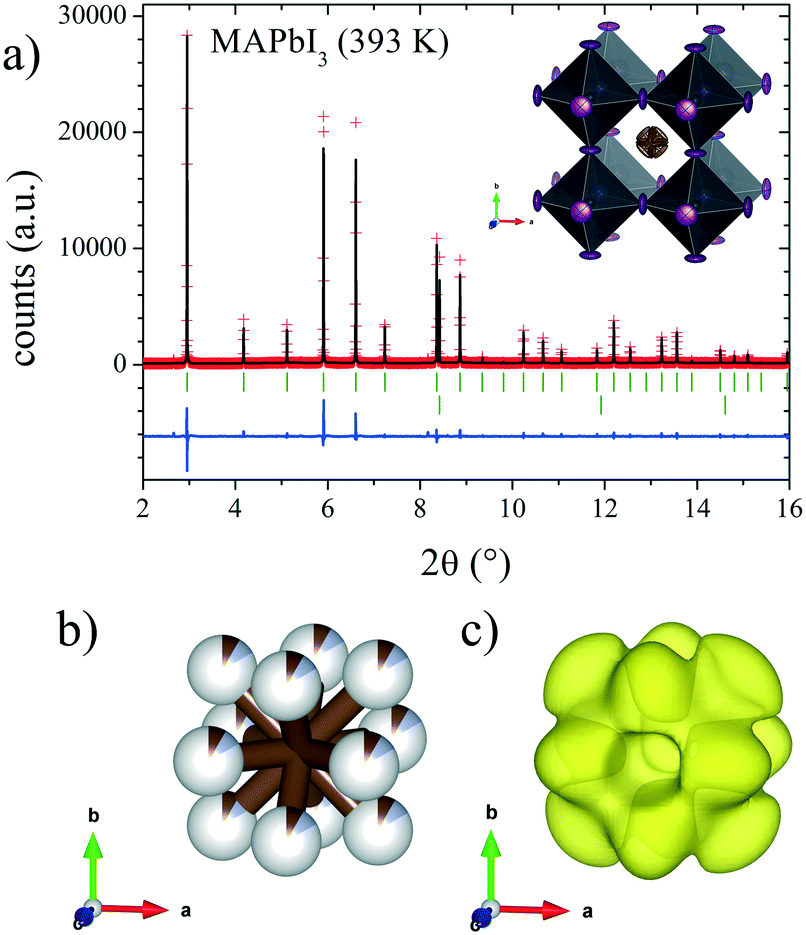 | ||
| Fig. 6 a) SXRD profile after Rietveld refinement at 393 K and schematic view of MAPbI3 (inset) at 393 K in the Pmm space group. b) and c) Detailed views of C/N positions and DFM density, respectively. | ||
MAPbI2Br
In contrast with MAPbI3, MAPbI2Br crystallizes at RT in the cubic phase with the space group Pmm, in the same way as described in a previous work for MAPbBr3 perovskite.29 Inspired by the behavior of this tribromide perovskite, which experiences two consecutive phase transitions from cubic Pmm to tetragonal I4/mcm (≈235 K) and then to orthorhombic Pnma (≈150 K),29,30 we additionally collected a SXRD pattern at 120 K for MAPbI2Br, observing that it also exhibits a cubic symmetry. In fact, the absence of transitions at low temperatures was also observed recently in mixed halide MAPb(Br1−xClx)3 perovskites where the anion disorder prevents the occurrence of phase transitions.8
In the present case, the DFM also was calculated to unveil the MA delocalization. As illustrated in Fig. 7, the isosurface density at 3e− reveals that the MA unit is aligned along the [111] direction at both RT and low temperature (120 K). Also, the electron density presents a particular triangular shape that is magnified at RT. This fact can be understood considering the H-bond interactions existing in these compounds. In the [111] delocalization, the three hydrogen atoms of MA units interact alike with the three halides that are in the face of a single PbX6 octahedron. However, taking into account the intrinsic halide disorder present in MAPbI2Br, it is expected that the face of the octahedron will not be symmetric, in average, inducing deviations towards some corners of the triangles. This is not unlikely, considering that 44.4% of the triangular faces are composed of two I and one Br, where the N–H⋯Br bonds will be shorter than the N–H⋯I distances. Fig. 8 shows the final refinements from the 298 and 120 K SXRD patterns, and Tables 2 and S3† list the main crystallographic data. The inset in Fig. 8b shows a schematic view of the obtained crystal structure.
 | ||
| Fig. 7 Schematic view of the PbX6 (X = I0.66Br0.33) framework (a) and electron density at RT (b) and (d) and at 120 K (c) and (e). | ||
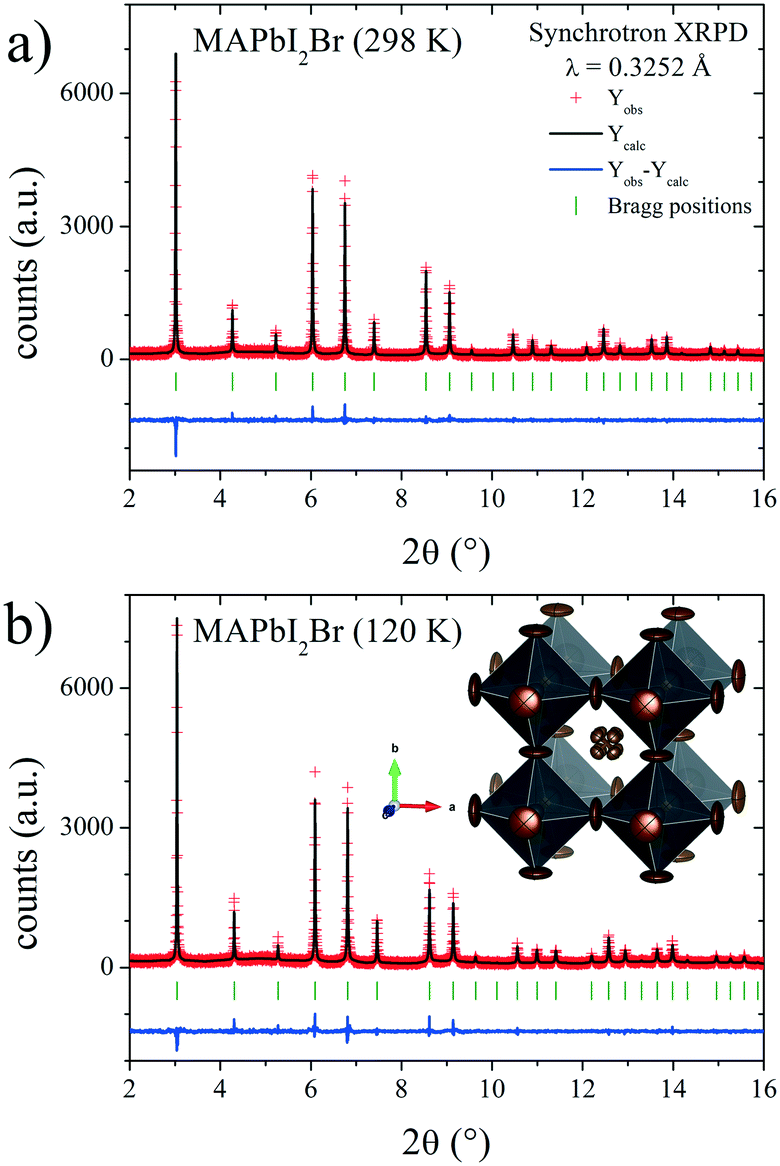 | ||
| Fig. 8 SXRD profiles after Rietveld refinement of MAPbI2Br at a) RT and b) 120 K. Inset in b): Schematic view of MAPbI2Br at RT. | ||
m) from SXRPD at RT. a = 6.17605(9) Å and V = 235.577(6) Å3
| x | y | z | U iso*/Ueq | Occ. | |
|---|---|---|---|---|---|
| Pb1 | 0 | 0 | 0 | 0.050(1) | 1 |
| Br1 | 0.5 | 0 | 0 | 0.122(3) | 0.37(3) |
| I1 | 0.5 | 0 | 0 | 0.122(3) | 0.63(3) |
| C1 | 0.569(3) | 0.569(3) | 0.569(3) | 0.03(2) | 0.12 |
| N1 | 0.569(3) | 0.569(3) | 0.569(3) | 0.03(2) | 0.12 |
| Atomic displacement parameters (Å2) | ||||||
|---|---|---|---|---|---|---|
| U 11 | U 22 | U 33 | U 12 | U 13 | U 23 | |
| R p = 11.1%, Rwp = 14.2%, χ2 = 1.02, RBragg = 10.7%. | ||||||
| Pb1 | 0.050(1) | 0.050(1) | 0.050(1) | 0.00000 | 0.00000 | 0.00000 |
| Br/I | 0.026(3) | 0.169(4) | 0.169(4) | 0.00000 | 0.00000 | 0.00000 |
| C1 | 0.03(2) | 0.03(2) | 0.03(2) | −0.01(1) | −0.01(1) | −0.01(1) |
| N1 | 0.03(2) | 0.03(2) | 0.03(2) | −0.01(1) | −0.01(1) | −0.01(1) |
Considering the used synthesis method, the microstructural behavior is another important aspect, which was considered in the Rietveld refinements. From this analysis, the average particle size is 176.3(3) nm and 84.4(1) nm for MAPbI3 and MAPbI2Br, respectively.
Optical band gap by UV-vis spectroscopy
Fig. 9 shows the optical absorption coefficient related to the Kubelka–Munk function (F(R) = a = (1 − R)2/2R, R being the reflectance of each sample) versus wavelength in eV, for both MAPbI3 (red line) and MAPbI2Br (blue line). The band gap for each perovskite has been calculated by extrapolating the linear region to the abscissa. The value obtained for MAPbI3 (∼1.6 eV) is typical for this material. The blue shift observed for the perovskite MAPbI2Br (∼1.75 eV) is also in accordance with data reported in the literature.31–33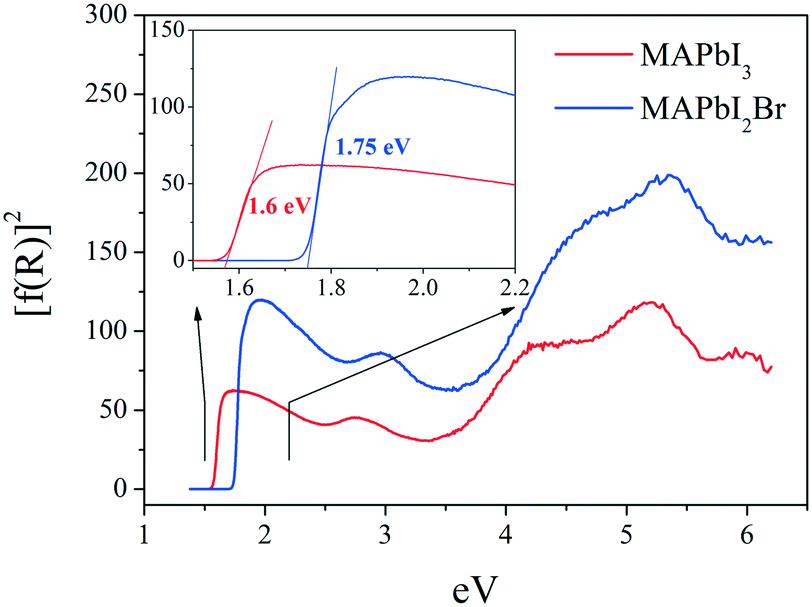 | ||
| Fig. 9 UV-vis absorption spectra for MAPbI3 (red line) and MAPbI3Br (blue line). | ||
Microstructure by scanning microscopy (SEM)
SEM images were obtained to understand the effect of the morphology of the crystals on the stability of the perovskites obtained by ball milling. Fig. 10 shows some typical views of the as-prepared MAPbI3 and MAPbI2Br. From a mechanosynthesis process, involving the collision of high energy ZrO2 balls against the specimens, one would expect a highly disaggregated product, formed by amorphous particles. However, surprisingly, we can observe a heterogeneous picture where quite large particles with sharp edges and large surfaces are mixed with smaller fragments with undefined shapes. This scenario illustrates that the growth of large microcrystals (as large as 20 microns in some edges) is not perturbed by the dynamic motion of the reactants and ZrO2 balls; the obtained images, after 4 hours of reaction, would suggest that the large particles grow at the expense of the remaining fine powder.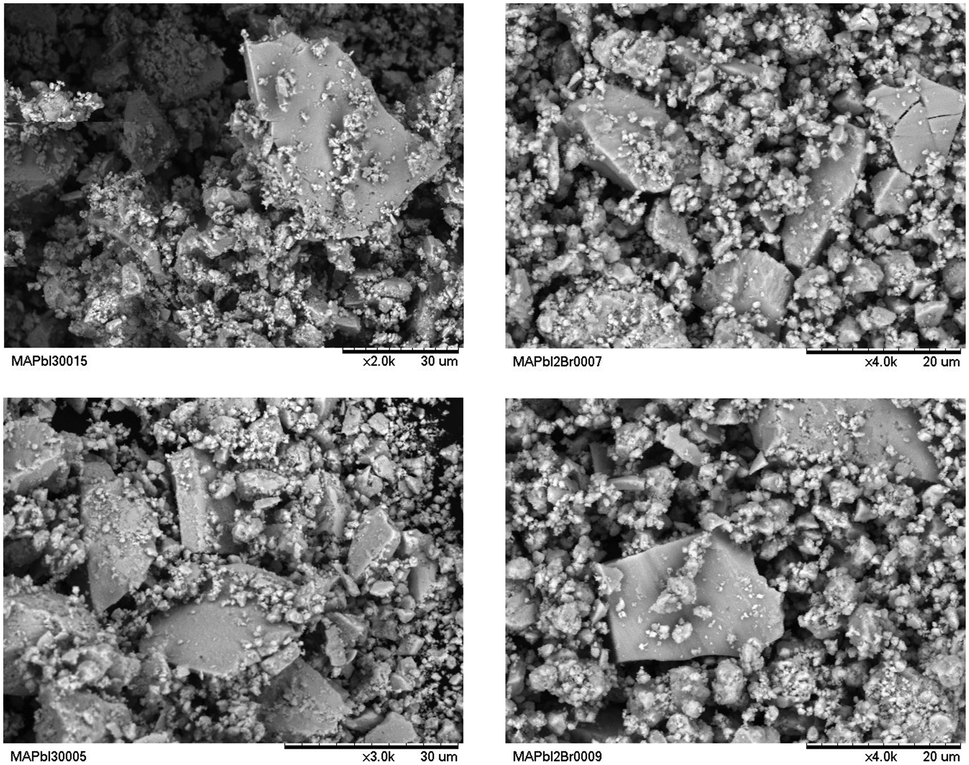 | ||
| Fig. 10 SEM images of MAPbI3 (left) and MAPbI2Br (right) powders. | ||
The reasonable size of the obtained crystals account for the good crystallinity observed in the diffraction diagrams, endorsing this procedure as complementary to the standard solution chemistry, with the added advantage of dispensing with organic solvents that, as we will see, are probably the origin of the observed chemical instability of these perovskites.
The results from EDX analysis are shown in Fig. S1 and S2 and Table S4 (ESI†). For both samples, the EDX spectra show well defined peaks corresponding to lead, bromine and iodine. The obtained weight% of these elements (Table S3†) are in agreement with the nominal values.
Chemical stability
Fig. 11 shows two XRD patterns of MAPbI3 prepared after 6 months, either by ball milling or crystallization from DMF, stored at ambient conditions in laboratory air (typically 40% humidity). The pattern of the sample obtained by mechanosynthesis is unaltered, with respect to the pristine pattern collected immediately after preparation (Fig. 1a); differences in peak intensities arise from preferred orientation effects. By contrast, in the XRD pattern of a sample of the same composition prepared by the standard procedure from a solution in DMF starting from MAI and PbI2, the peaks of the perovskite are accompanied by significant amounts of impurities, mainly MAI and PbI2. Even visually, after some weeks since it was synthesized, it is observed that the characteristic black color is decorated by grey particles, appearing inhomogeneous even at the macroscopic scale.34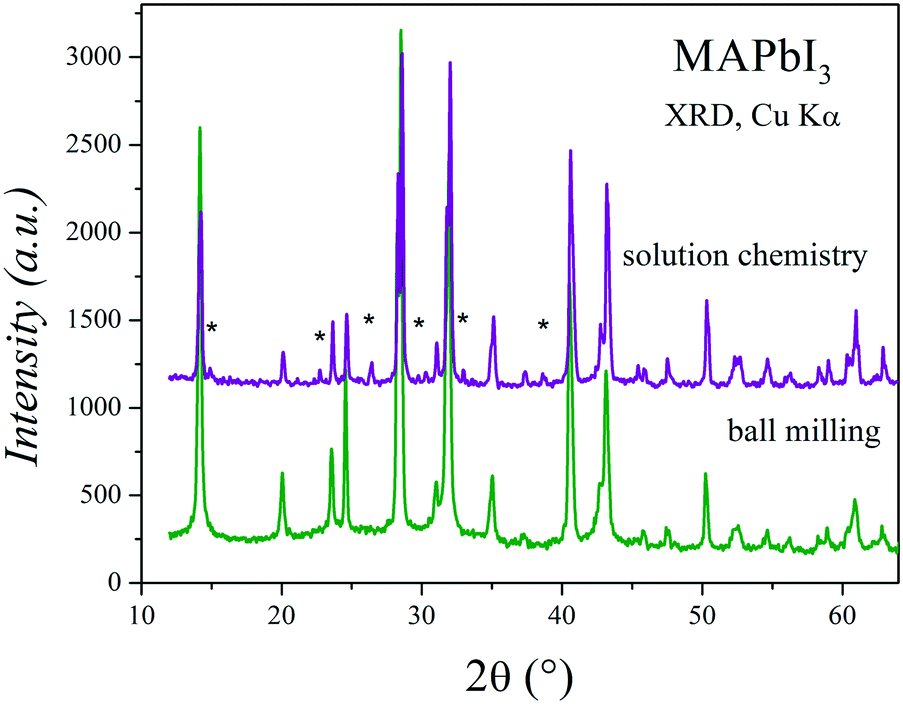 | ||
| Fig. 11 Two XRD patterns of MAPbI3 prepared after 6 months, either by crystallization from DFM (solution chemistry) or ball milling. | ||
The absence of crystalline impurities and high yield of the mechanochemical synthesis of MAPbI3 in comparison to those of other methods (such as reaction in solution) have been also reported in the literature, even for the samples that are recently prepared.12
In fact, the water degradation mechanism of MAPbI3 was previously studied by different authors. Niu et al.35 reported that MAPbI3 degrades into PbI2 and MAI in the presence of H2O, where MAI further decomposes into CH3NH2(aq) and HI(aq). Frost et al.36 proposed two reversible reactions, where the combination of H2O and nMAPbI3 leads to the formation of CH3NH2 and [(CH3NH3+)n−1(CH3NH2)nPbI3][H2O], the latter further degrading into [(CH3NH3+)PbI3]n−1, HI, PbI2 and H2O. On the other hand, Habisreutinger et al.37 speculated that the weak hydrogen bonds allow MAI to escape from the crystal lattice, which eventually leads to the formation of PbI2. Another possible reason mentioned for the instability of MAPbI3 could be related to the interaction of residual traces of DMF. Thus, the hydrolysis of DMF traces could form dimethylammonium formate, compromising the ability of Pb2+ and MA+ to stabilize the perovskite structure.38
Regardless of the mechanism, it seems that the degradation occurs when the MA units are in contact with the environment. Moreover, it is well known that, during the mechanical milling, a lot of high-energy collisions are induced, which generate high temperatures locally for short periods of time. This process drives the reaction; however, this also generates a high disorder in the crystal surface and probably defects of MA+. Considering these facts, it is possible to infer that the dry mechanochemical synthesis produces a surface deficient in MA+, which slows down the MAPbI3 decomposition similarly to a “passivation layer”. Moreover, it seems it is not only a surface behaviour, but the mechanochemical synthesis could also affect the bulk crystal features. As stated previously, the inorganic framework presents a conspicuously lower tilting effect than those observed in samples prepared by wet methods; an additional difference arises since the tetragonal structure is stable down to 140 K. The smaller octahedral rotation suggests an increased tolerance factor of the perovskite, which is a known consequence of the partial deficiency of MA at the A positions of the ABX3 perovskite, and therefore an improved structural stability, also accounting for the absence of structural transitions down to 140 K. Both effects and, finally, the chemical stability increase, which can be attributed to both surface and bulk crystal features or a combination of them.
Conclusions
Two hybrid perovskites of the family MAPbX3 have been successfully prepared by mechanochemical synthesis in an inert atmosphere. They have been demonstrated to exhibit superior chemical stability towards humidity compared to specimens prepared by solution chemistry. Despite the dynamic process inherent in the ball-milling process, well-crystallized specimens have been obtained, which allowed a high resolution SXRD study to be carried out. MAPbI3 is better defined in the tetragonal I4/mcm space group between RT and 140 K; difference Fourier synthesis allowed us to determine the orientation of MA+ units, which evolves with temperature even if the symmetry does not change. At 393 K, this triiodide perovskite becomes cubic with the space group Pmm. Several conspicuous structural features of MAPbI3 may be related to a better stability, probably originating from the elimination of MA+ during the ball milling process; these include the absence of an additional phase transition to orthorhombic symmetry at 140 K, distinct tilting angles and MA conformation at different temperatures. Regarding MAPbI2Br, it is cubic in the 120–298 K range, suggesting that the anion disorder prevents the occurrence of phase transitions. In this case, the MA unit is aligned along the [111] direction in all the temperature range.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support given by the Spanish Ministry of Economy and Competitiveness (MAT2017-84496-R) and KIC RAW MATERIALS from the European Union (OPTNEWOPT project). C. A. L. acknowledges ANPCyT and UNSL, Argentina, for the financial support (projects PICT2017-1842 and PROICO 2-2016, respectively).References
- S. Ananthakumar and S. Moorthy Babu, Synth. Met., 2018, 246, 64–95 CrossRef CAS.
- A. Dubey, N. Adhikari, S. Mabrouk, F. Wu, K. Chen, S. Yang and Q. Qiao, J. Mater. Chem. A, 2018, 6, 2406–2431 RSC.
- W. Rehman, D. P. McMeekin, J. B. Patel, R. L. Milot, M. B. Johnston, H. J. Snaith and L. M. Herz, Energy Environ. Sci., 2017, 10, 361–369 RSC.
- S. Kazim, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2014, 53, 2812–2824 CrossRef CAS PubMed.
- A. Sadhanala, F. Deschler, T. H. Thomas, S. E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 2501–2505 CrossRef CAS PubMed.
- B. Park, S. M. Jain, X. Zhang, A. Hagfeldt, G. Boschloo and T. Edvinsson, ACS Nano, 2015, 9, 2088–2101 CrossRef CAS PubMed.
- E. M. Talbert, H. F. Zarick, N. J. Orfield, W. Li, W. R. Erwin, Z. R. DeBra, K. R. Reid, C. P. McDonald, J. R. McBride, J. Valentine, S. J. Rosenthal and R. Bardhan, RSC Adv., 2016, 6, 86947–86954 RSC.
- M. C. Alvarez-Galván, J. A. Alonso, C. A. López, E. López-Linares, C. Contreras, M. J. Lázaro, F. Fauth and M. V. Martínez-Huerta, Cryst. Growth Des., 2019, 19, 918–924 CrossRef.
- Z. Fang and Z. Yi, Chem. Phys. Lett., 2017, 687, 19–22 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- A. D. Jodlowski, A. Yépez, R. Luque, L. Camacho and G. de Miguel, Angew. Chem., Int. Ed., 2016, 55, 14972–14977 CrossRef CAS PubMed.
- A. Jana, M. Mittal, A. Singla and S. Sapra, Chem. Commun., 2017, 53, 3046–3049 RSC.
- P. Sadhukhan, S. Kundu, A. Roy, A. Ray, P. Maji, H. Dutta, S. K. Pradhan and S. Das, Cryst. Growth Des., 2018, 18, 3428–3432 CrossRef CAS.
- D. Prochowicz, M. Franckevičius, A. M. Cieślak, S. M. Zakeeruddin, M. Grätzel and J. Lewiński, J. Mater. Chem. A, 2015, 3, 20772–20777 RSC.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628 RSC.
- F. Fauth, R. Boer, F. Gil-Ortiz, C. Popescu, O. Vallcorba, I. Peral, D. Fullà, J. Benach and J. Juanhuix, Eur. Phys. J. Plus, 2015, 130, 160 CrossRef.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS.
- Y. Kawamura, H. Mashiyama and K. Hasebe, J. Phys. Soc. Jpn., 2002, 71, 1694–1697 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- M. T. Weller, O. J. Weber, P. F. Henry, A. M. Di Pumpo and T. C. Hansen, Chem. Commun., 2015, 51, 4180–4183 RSC.
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1990, 51, 1383–1395 CrossRef CAS.
- S. Govinda, P. Mahale, B. P. Kore, S. Mukherjee, M. S. Pavan, C. De, S. Ghara, A. Sundaresan, A. Pandey, T. N. Guru Row and D. D. Sarma, J. Phys. Chem. Lett., 2016, 7, 2412–2419 CrossRef PubMed.
- K. Frohna, T. Deshpande, J. Harter, W. Peng, B. A. Barker, J. B. Neaton, S. G. Louie, O. M. Bakr, D. Hsieh and M. Bernardi, Nat. Commun., 2018, 9, 1829 CrossRef PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- C. A. López, M. V. Martínez-Huerta, M. C. Alvarez-Galván, P. Kayser, P. Gant, A. Castellanos-Gomez, M. T. Fernández-Díaz, F. Fauth and J. A. Alonso, Inorg. Chem., 2017, 56, 14214–14219 CrossRef PubMed.
- I. P. Swainson, R. P. Hammond, C. Soullière, O. Knop and W. Massa, J. Solid State Chem., 2003, 176, 97–104 CrossRef CAS.
- Y. Zhao and K. Zhu, J. Am. Chem. Soc., 2014, 136, 12241–12244 CrossRef CAS PubMed.
- K. Cao, J. Cui, H. Zhang, H. Li, J. Song, Y. Shen, Y. Cheng and M. Wang, J. Mater. Chem. A, 2015, 3, 9116–9122 RSC.
- P. F. Ndione, Z. Li and K. Zhu, J. Mater. Chem. C, 2016, 4, 7775–7782 RSC.
- X. Zhao and N.-G. Park, Photonics, 2015, 2, 1139–1151 CrossRef CAS.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 RSC.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- B. Dou, L. M. Wheeler, J. A. Christians, D. T. Moore, S. P. Harvey, J. J. Berry, F. S. Barnes, S. E. Shaheen and M. F. A. M. van Hest, ACS Energy Lett., 2018, 3, 979–985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce01461f |
| This journal is © The Royal Society of Chemistry 2020 |