
Influence of ortho- and meta-substituted N-heterocyclic dications towards the self-assembly of p-sulfonatocalix[4]arene†
Sze Le
Loh
a,
Alexandre N.
Sobolev
b,
Siew Huah
Lim
c and
Irene
Ling
 *a
*a
aSchool of Science, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 47500 Selangor, Malaysia. E-mail: ireneling@monash.edu
bChemistry Department, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia
cSchool of Molecular Sciences and CMCA, M310, The University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia
First published on 29th October 2019
Abstract
The interplay of organic dications bearing various n-alkylimidazolium termini substituted at different positions on a bis(methylene)phenyl ring and p-sulfonatocalix[4]arene, along with tetraphenylphosphonium cations and hydrated lanthanides (Ce3+, Er3+ and Gd3+), afforded various self-assembled solid-state structures. The host–guest binding behaviour of the dications and p-sulfonatocalix[4]arene was carefully investigated by crystallographic studies including their interactions in the solution state, but in the absence of phosphonium and lanthanide cations via1H NMR experiments.
Introduction
Macrocyclic molecules have gained attention from chemists and biochemists notably in the field of supramolecular chemistry owing to their excellent molecular recognition via host–guest interactions and their profound role as binding site models in biological systems.1–5 Calix[n]arenes are one of the most studied macrocyclic molecules notably in the areas of supramolecular chemistry and crystal engineering due to their flexible chemical modification feasibility by attaching different functional groups comprising a broad range of donor atoms for complexation, and variable cavity sizes and molecular shapes. Water-soluble calix[n]arene, particularly p-sulfonatocalix[4]arene, 1, in its most stable cone conformation, has garnered attention for its importance in biological sciences, material sciences and medicinal chemistry due to its biocompatibility and strong binding affinity towards numerous bioactive molecules6–8 including amino acids, enzymes, peptides and proteins9,10 as well as biological substrates such as drugs, imaging agents and genes.11One of the initial successes in rationalising host–guest interactions based on 1 and N-heterocyclic molecules was found during the investigation of pyridine and N-oxide in the solid state.12–16 Deep pyridine ring inclusion with the N and O atoms directed away from the cavity is shown to be favourable from the judicious electronic profile of a pyridine N-oxide molecule to form intermolecular C–H⋯π interaction. It was in this context, and in continuation of related studies, a series of closely related monoimidazolium ions that featured in ionic liquids, differing only in the nature of the n-alkyl chain substituent in the 3-position of the imidazole ring, have been investigated by crystallographic studies.17–19 A common host–guest pair was observed in all complexes where the heterocyclic ring is persistently taken up by the calixarene with the aliphatic tail projecting away from the hydrophilic segment of the macrocyclic molecule. Investigations of the host–guest interplay of 1 with N-heterocyclic molecules continued thereafter, integrating a series of para-substituted dications bearing imidazolium substituents, with varying conclusions.20–23 The inclusion of para-substituted dications bearing either the methyl or n-butyl chain on the imidazolium ring often yields a molecular capsule arrangement, end-capped by two opposing calixarenes and neatly packed into bilayers.24 In selected cases, for the same para-substituted dication as well as for relatively longer alkyl chain dications, partial inclusion was achieved where only one imidazolium moiety is drawn into the calixarene cavity with the other directed away from the cavity without perturbing the overall bilayer arrangement.22
In the present work, we aim to investigate the effect of guest substituent positions towards the self-aggregation of 1 (Scheme 1) in the presence of ortho- and meta-disubstituted methyl- and butylimidazoliums. A hierarchical strategy was attempted where multi-component complexes comprising ortho- or meta-substituted methyl- or n-butylimidazolium dicationic salts and 1 either in the presence or absence of both monophosphonium and lanthanide cations were prepared (ESI†). We have successfully crystallised nine complexes yielded from the slow evaporation of 1 with 3,3′-(1,2-phenylenebis(methylene))bis(1-methyl-1H-imidazol-3-ium) 2, 3,3′-(1,2-phenylenebis(methylene))bis(1-butyl-1H-imidazol-3-ium) 3, or 3,3′-(1,3-phenylenebis(methylene))bis(1-methyl-1H-imidazol-3-ium) 4, in the presence of lanthanide halide salts (cerium(III), erbium(III), or gadolinium(III)) either with or without tetraphenylphosphonium bromide, Ph4P+Br. Attempts to grow suitable single crystals of meta-substituted n-butylimidazolium dicationic salt in the presence of 1 under similar experimental conditions were not successful. Similarly, we did not succeed in growing crystals using other combinations and the corresponding combinatorial matrices are summarized in Table S1, ESI†.
 | ||
| Scheme 1 p-Sulfonatocalix[4]arene, 1. | ||
Results and discussion
The structural elucidation for nine different multi-component complexes involving 1 and 2, 3, or 4 along with the presence or absence of tetraphenylphosphonium cations and lanthanide cations (cerium(III), erbium(III) and gadolinium(III)) has been carefully conducted (Table 1). The availability of these closely related structures has enabled a systematic investigation on the influence of substitution positions at the bis(methylene)phenyl ring upon the supramolecular host–guest interplay based on various weak intermolecular interactions. The substituent effects, related to the positions and sizes, are shown to be marked in terms of the ways in which the molecules aggregate to form different host–guest assemblies. Additionally, steric considerations for these dications have proven critical in the control of the manner by which host, guest and other auxiliary molecules interact.| Complex | 1 | 2 | 3 | 4 | LnCl3·xH2O | PPh4+ |
|---|---|---|---|---|---|---|
| I | ✓ | ✓ | Gd | |||
| II | ✓ | ✓ | Ce | |||
| III | ✓ | ✓ | Er | |||
| IV | ✓ | ✓ | Ce | |||
| V | ✓ | ✓ | Ce | |||
| VI | ✓ | ✓ | Gd | |||
| VII | ✓ | ✓ | Er | |||
| VIII | ✓ | ✓ | Ce | ✓ | ||
| IX | ✓ | ✓ | Gd | ✓ |
Host–guest interplay
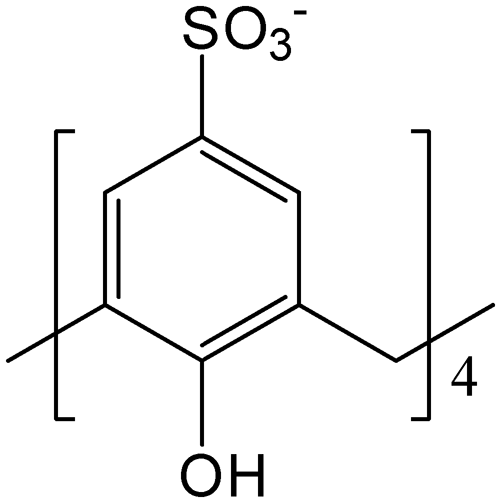
As anticipated from the structural compositions, significant non-classical hydrogen bonding is dominant and thus drives the stability of the overall supermolecule formation in the solid state (complexes I to IX).The inclusion behaviour involving ortho-disubstituted methylimidazolium, 2, in the presence of 1 (complex I) shows two different interplays within the same structure where the first type takes on a capsular configuration (approx. size = 14 Å) albeit slightly skewed where the bis(methylene)phenyl ring and one of the methylimidazolium moieties of 2 are capped by two independent calixarenes (Fig. 1). The integral part of the aforementioned supermolecule formation was attributed to evident intermolecular C–H⋯π(phenyl) interactions with C⋯π(phenyl) distances ranging from 3.43 to 3.67 Å along with extensive hydrogen bonding, particularly at the upper rim where the C–H⋯O distances range from 2.29 to 2.78 Å. Due to the CH2 group rotational limitation, the other imidazolium moiety is directed near the capsule seam and participates in hydrogen bonding with the sulfonate groups via the oxygen atoms, with C–H⋯O contacts ranging from 2.52 to 2.99 Å (corresponding C⋯O distances from 3.15 to 3.50 Å). The second type shows that only one imidazolium moiety is accommodated in the calixarene cavity, and the two components are involved in the C⋯π(phenyl) interaction with close contacts ranging from 3.11 to 3.91 Å and the C–H⋯O interaction with the sulfonate groups ranging from 2.23 to 2.99 Å (corresponding C⋯O distances from 3.15 to 3.43 Å). The bis(methylene)phenyl ring and imidazolium moiety that do not participate directly in the inclusion are involved in the π(phenyl)⋯π(phenyl) stacking and hydrogen bonding with neighbouring calixarenes with calculated π(phenyl)⋯π(phenyl) distances of 3.55 Å and 3.68 Å and C–H⋯O distances ranging from 2.48 to 2.93 Å (corresponding C⋯O distances from 3.34 to 3.78 Å), respectively, ultimately stabilising the overall cohesion in the crystal packing.
 | ||
| Fig. 1 The diversity of host–guest binding behaviour involving 1 and 2, 3 or 4 and resulting in the formation of different supermolecules from their interplay. | ||
For supermolecules involving 1 and 3 (half-populated in complex II while fully populated in complex III), the n-butyl group in 3 is considerably more flexible, therefore interplays with calixarene through multiple ways (Fig. 1). In complex II, the ortho-disubstituted butylimidazolium cation has its bis(methylene)phenyl ring and one of the n-butyl chains positioned in two independent calixarene cavities with the other n-butylimidazolium moiety directed away from the cavities, likewise in complex I. Interestingly, the bis(methylene)phenyl ring is not drawn deep into the cavity as opposed to complex I and it is in close proximity to the calixarene hydrophilic upper rim where the ring readily hydrogen bonds with the available oxygen atoms, with C–H⋯O contacts of 2.65 and 3.02 Å (corresponding C⋯O distances of 3.35 and 3.79 Å). Interestingly, the confined butylimidazolium ring has its n-butyl chain slightly folded in the calixarene cavity and this type of binding configuration has been reported previously involving the interplay of mono-butylimidazolium and calixarene.24 Calculated intermolecular interactions include C⋯π(phenyl) interactions at 3.52 Å and 3.70 Å and hydrogen bonding with C–H⋯O distances ranging from 2.58 to 2.91 Å (corresponding C⋯O distances from 3.37 to 3.52 Å). As for the non-confined butylimidazolium group, the fragment interacts strongly with the sulfonate groups at the seam where hydrogen bonding is the crucial intermolecular interaction and the C–H⋯O distances range from 2.45 to 2.74 Å (corresponding C⋯O distances from 3.14 to 3.43 Å).
Deep inclusion of the bis(methylene)phenyl ring into the calixarene cavity is observed in complex III in which C⋯π(phenyl) interaction (distances of 3.49 Å and 3.57 Å) and C–H⋯O interaction (close distance of 2.72 Å; corresponding C⋯O distance of 3.467 Å) are the dominant intermolecular interactions stabilising the overall self-aggregation. It is noteworthy that such a host–guest arrangement prohibits the end-capping of both the butylimidazolium termini and the n-butyl chains, thus these fragments are directed away from the calixarene cavity and are in closer positions with the calixarene sulfonate groups, with C–H⋯O contacts from 2.24 to 2.92 Å (corresponding C⋯O distances from 3.05 to 3.48 Å). Other close contacts involving the n-butyl chains and neighbouring calixarene phenyl rings and methylene bridges include C⋯π(phenyl) and C⋯C distances of 3.86 Å and 3.85 to 4.14 Å, respectively.
Different ways of self-aggregation in the solid state involving 1 and meta-disubstituted methylimidazolium, 4, were observed despite two of the host–guest complexes showed similar inclusion behaviour to that in complex I (Fig. 1). Supermolecules in complexes IV and VIII have the bis(methylene)phenyl ring and one imidazolium moiety of 4 being confined within two independent calixarenes affording a skewed capsular configuration (approx. size = 14 Å for both complexes). Owing to the molecule's geometry, the bis(methylene)phenyl ring is positioned in a slantwise manner but not deep in the calixarene cavity and is involved in the C⋯π(phenyl) interaction through the calixarene phenyl rings, with the shortest contacts found to be 3.74 Å and 3.24 Å and also assisted with additional stabilisation by C–H⋯O hydrogen bonding involving the methylene bridge with the calixarene sulfonate group, with the shortest contacts of 2.68 Å and 2.56 Å (corresponding C⋯O distances of 3.49 Å and 3.25 Å) for complexes IV and VIII, respectively. One of the methylimidazolium groups fits deeply in an adjacent calixarene cavity stabilised through hydrogen bonding to the calixarene phenyl rings and sulfonate groups, with C⋯π(phenyl) contacts ranging from 3.49 to 3.74 Å and C–H⋯O short contacts ranging from 2.42 to 3.08 Å (corresponding C⋯O distances from 3.03 to 3.71 Å) for both complexes IV and VIII. Likewise in complex I, the non-confined methylimidazolium group points towards the capsule seam and participates in hydrogen bonding with the surrounding sulfonate groups with calculated C–H⋯O short contacts ranging from 2.32 to 2.91 Å (corresponding C⋯O distances from 3.06 to 3.45 Å). A different binding mode between 1 and 4 in complexes IV to VII is noted, where only one of two methylimidazolium moieties is successfully drawn into the calixarene cavity, with close C⋯π(phenyl) distances from 3.48 to 3.61 Å and C–H⋯O contacts from 2.50 to 2.96 Å (corresponding C⋯O distances from 3.34 to 3.54 Å). The non-confined methylimidazolium group is directed to adjacent calixarene and involved in π-stacking with the calixarene phenyl ring (π(phenyl) ⋯π(phenyl) distances from 3.57 to 3.59 Å) and simultaneously hydrogen bonds to the neighbouring sulfonate groups, with C–H⋯O contacts from 2.39 to 2.65 Å (corresponding C⋯O distances from 3.22 to 3.30 Å).
A common host–guest inclusion configuration to supermolecules constructed from para-disubstituted alkylimidazoliums and calixarenes in previously reported structures is observed in complex IX where both methylimidazolium termini are selectively drawn into the calixarene cavity forming a capsule arrangement albeit skewed (approx. size = 16 Å). In one end, the methyl is directed into the cavity and interacts through the C⋯π(phenyl) interaction with four calixarene phenyl rings, with C⋯π(phenyl) distances from 3.50 to 3.72 Å, whilst the other has the imidazolium ring interacting with four calixarene phenyl rings with C⋯π(phenyl) contacts from 3.32 to 3.54 Å. The host–guest interplay is further stabilised by hydrogen bonding from the methylene bridge, bis(methylene)phenyl ring and imidazolium moiety to the sulfonate groups with calculated C–H⋯O distances ranging from 2.28 to 2.95 Å.
Self-assembly
The majority of the structures (except for complexes I, IV and IX) adopt the bilayer arrangement although the topologies of the included guest molecule vary. All the complexes crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (except for complex IX where it crystallised in the monoclinic, space group P21/c) and lanthanide ions were found to exert major influence on the overall crystal packing. The calixarenes in all the complexes adopt the expected cone conformation (dihedral angles for all the complexes are summarized in the ESI†).
(except for complex IX where it crystallised in the monoclinic, space group P21/c) and lanthanide ions were found to exert major influence on the overall crystal packing. The calixarenes in all the complexes adopt the expected cone conformation (dihedral angles for all the complexes are summarized in the ESI†).
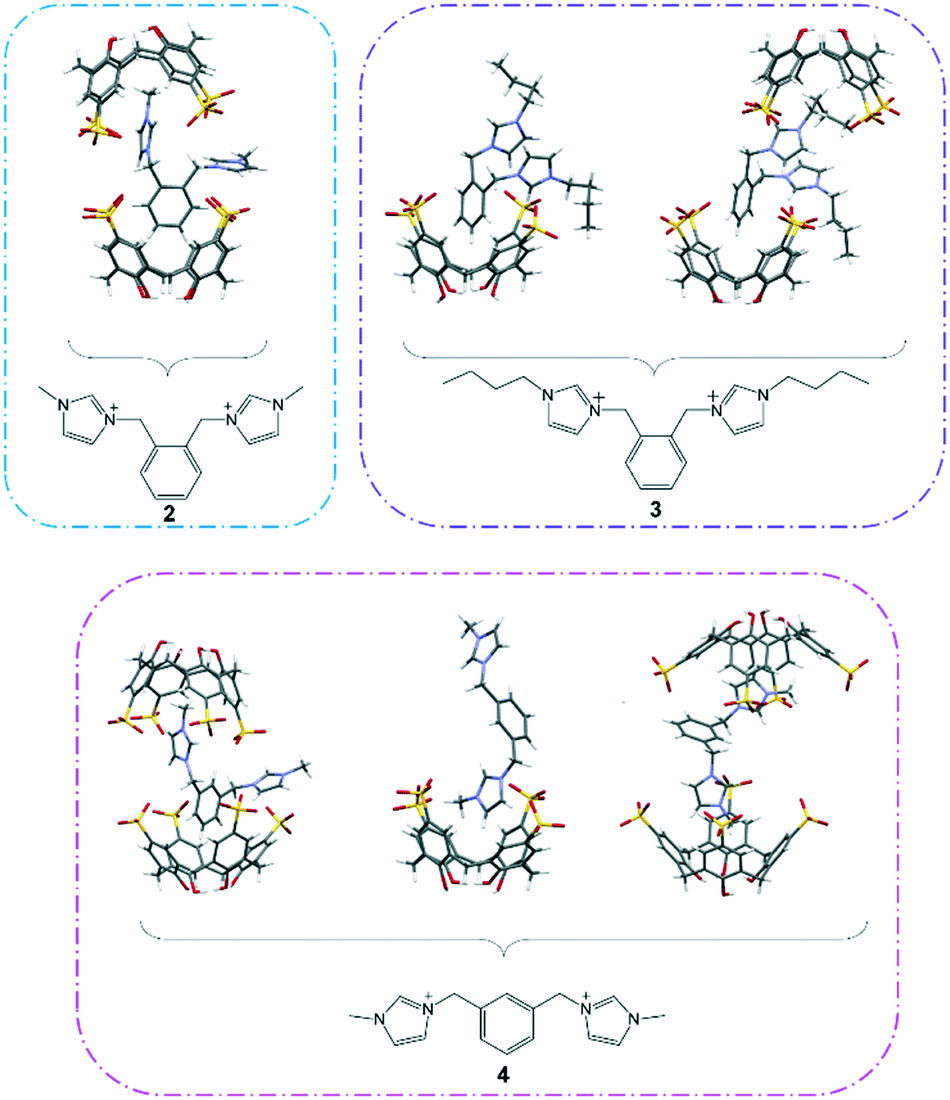
The extended structure in complex I is intricate and does not feature the common bilayer arrangement of the calixarene with well separated hydrophobic and hydrophilic layers. Rather, it is built from regularly arranged molecular capsules and each of the capsules is separated by layers of divergent back-to-back calixarenes (Fig. 2). Different coordination environments were identified for the partially occupied gadolinium(III) ions present in the complex, either directly coordinating to a sulfonate group, or forming a secondary coordination sphere with adjacent calixarene molecules. Two gadolinium(III) ions are in close positions to the sulfonate groups located at the seam of the capsule forming secondary coordination spheres to the calixarene upper rims (Gd–O⋯O–S distances ranging from 2.360(2) to 2.973(2) Å). The other two heteroleptic gadolinium(III) ions directly bound to sulfonate groups (Gd–O = 2.224(7) Å and 2.326(9) Å) with one of the metal ions have an incomplete coordination sphere. A sodium ion bridges a capsule and an independent calixarene via direct coordination through the sulfonate group from each calixarene, with Na–O contacts of 2.334(9) and 2.380(1) Å. 2 also contributes to the overall cohesion of the self-assembly where the bis(methylene)phenyl ring and imidazolium moiety that do not engage in the host–guest inclusion are involved in π(phenyl)⋯π(phenyl) stacking and hydrogen bonding to adjacent calixarene phenyl rings and oxygen atoms with π(phenyl)⋯π(phenyl) distances from 3.55 to 3.68 Å and C–H⋯O distances from 2.488 to 2.93 Å (corresponding C⋯O distances from 3.34 to 3.78 Å).
 | ||
| Fig. 2 Crystal packing of complex I with partial space filling showing the skewed capsule (i) while the dark blue color highlights the layer of calixarenes in divergent back-to-back arrays (ii). | ||
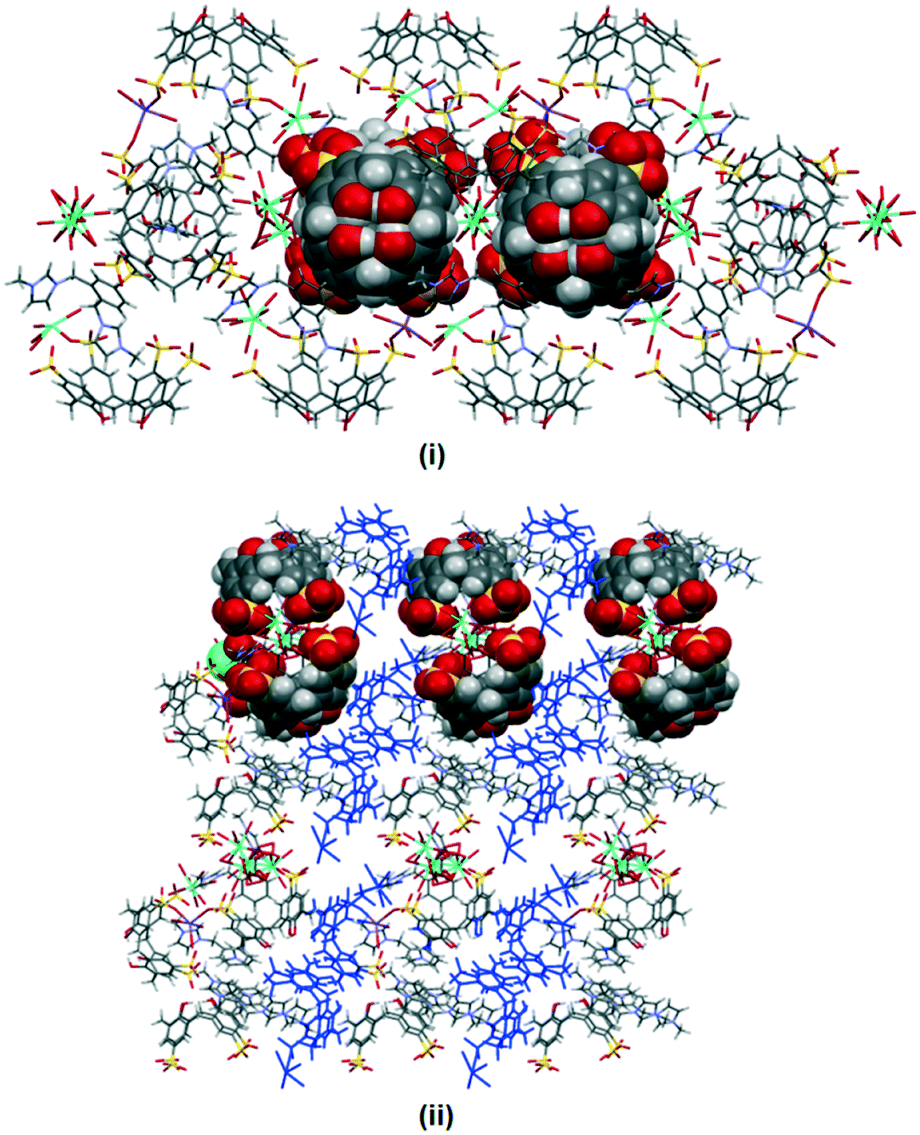
The crystal packing in complex II is analogous to the typical bilayer arrangement that is consistently found in calixarene and imidazolium molecules (Fig. 3(i)). The bilayer arrangement is essentially built from molecular capsules that are coordinated by cerium(III) ions through a secondary coordination sphere. It is noteworthy that each of the skewed capsule (approx. size = 17 Å) is surrounded by four fully populated nine-coordinate cerium(III) ions at the seam with Ce–O⋯O–S distances ranging from 2.719(3) to 2.838(2) Å. Each calixarene also has its sulfonate groups directly linked to a sodium cation (Na–O distance of 2.939(4) Å) with the remaining coordination sphere of sodium ion being completed by two water molecules (Na–O distances of 2.442(6) Å and 2.476(5) Å).
Replacing cerium(III) ions with erbium(III) ions afforded complex III and the bilayers are consolidated by the coordination of the eight coordinate heteroleptic erbium(III) ions (Er–O–S = 2.375(3) Å, Er–O(water) distances from 2.290(2) to 2.369(3) Å) through secondary coordination to the calixarenes at the hydrophilic layer, with Er–O⋯O–S from 2.621(2) to 2.819(3) Å. Further stabilisation to the bilayer is provided by sodium ions through a direct coordination of the half-populated sodium ion, at the crystallographic inversion center, that bridges a pair of oxygen atoms from two opposing calixarenes (Na–O distances of 2.328(6) and 2.640(7) Å) that have a −3 charge. The non-confined butylimidazolium terminus also interacts with the surrounding calixarene molecules with the detailed short contacts as discussed above (Fig. 3(ii)).
 | ||
| Fig. 3 Bilayer arrangement of complexes II (i) and III (ii) with partial space filling showing 3 contained within the calixarenes and the interstices. | ||
The supramolecular architecture in complex IV is similar to that in complex I. Molecular capsules are arranged in a regular manner and are separated by layers of divergent back-to-back calixarenes where all calixarenes have a −2 charge. The capsule is stabilized by two cerium(III) ions, one heteroleptic with a Ce–O distance of 2.514(8) Å, along with eight water molecules completing the coordination sphere (Ce–O distances ranging from 2.442(10) to 2.570(9) Å) while the other homoleptic forms secondary coordination interaction with the calixarene upper rims. The calixarenes that are at right angles to each capsule have a cerium(III) ion that directly coordinates to a calixarene sulfonate group with a Ce–O distance of 2.518(7) Å and further interacts with calixarene sulfonate groups through hydrogen bonding, with Ce–O⋯O–S distances ranging from 2.701(1) to 2.989(2) Å. Sodium ions are present in the final structure and each ion coordinates to an oxygen atom of one calixarene sulfonate group with a Na–O distance of 2.334(9) Å and the remaining coordination sphere is filled by four water molecules (Na–O distances ranging from 2.321(8) to 2.397(8) Å). The guest molecule also participates in the overall assembly where the bis(methylene)phenyl ring and independent imidazolium group of 4 which are not involved in the host–guest interplay participate in the C–H⋯O interactions with calixarene sulfonate groups, with close distances ranging from 2.52 to 2.90 Å (corresponding C⋯O distances from 3.11 to 3.66 Å) and C–H⋯H–C contacts of 2.53 Å and 2.87 Å with the calixarene methylene bridges (corresponding C⋯C are 3.13 Å and 3.32 Å).
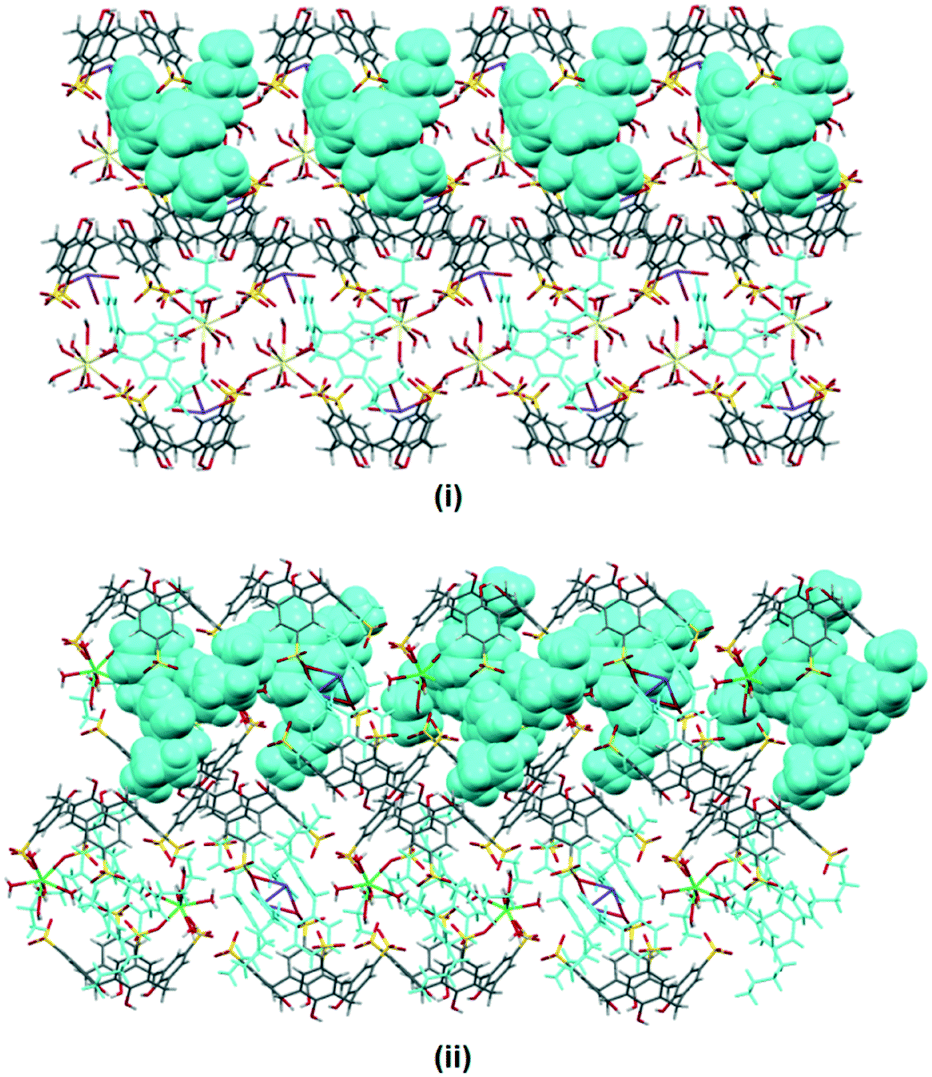
Synthesis of 1 and 4 along with cerium(III) ions also yields a polymorph that has a different crystal packing, complexes IV and V. Complex V has a different asymmetric unit compared to complex IV and the extended structure is built from a distorted bilayer arrangement which is coordinated by cerium(III) ions through secondary coordination, with Ce–O⋯O–S distances ranging from 2.690(1) to 2.984(9) Å (Fig. 4). Not all the calixarene cavities have the guest molecule included as the others have aquated cerium(III) ions located at the cavity which presumably inhibit any guest inclusion, with close Ce–O⋯O–S distances recorded from 2.76 to 2.89 Å. Each supermolecule that has 4 included in the cavity interacts via π(phenyl)⋯π(phenyl) stacking through bis(methylene)phenyl, with a distance of 3.59 Å which promotes additional stability to the overall self-aggregation. Complexes VI and VII are isostructural to complex V, except the metal ions included in the crystal structure are gadolinium(III) and erbium(III) ions, respectively.
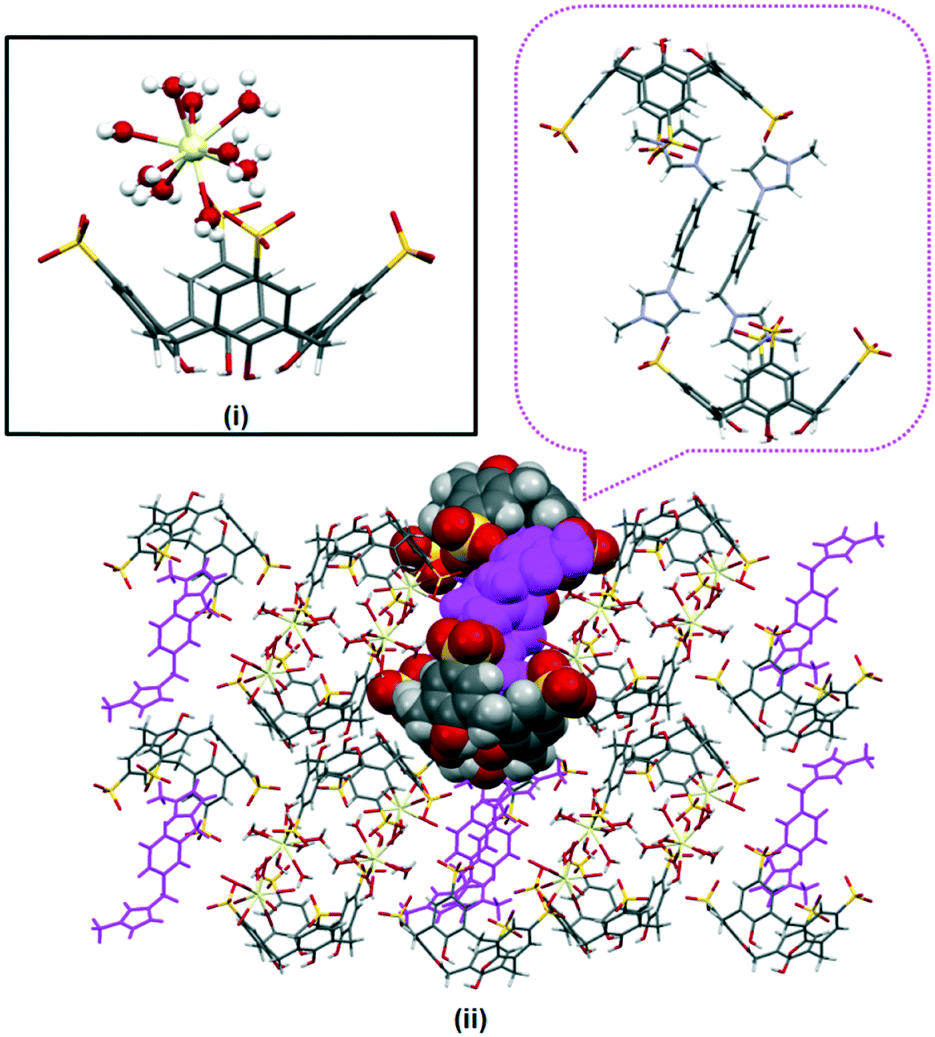 | ||
| Fig. 4 Bilayer arrangement of complex V with partial space filling (pink) showing the inclusion complex and each supermolecule is connected via π(phenyl)⋯π(phenyl) stacking (ii). (Inset (i)) Aquated cerium(III) ions that are positioned at the calixarene upper rim avert successful guest inclusion. | ||
Complex VIII was prepared under similar conditions to complex V but with the addition of an auxiliary component, the tetraphenylphosphonium cation. The extended structure is constructed from an arrayed skewed molecular capsule that has three nine-coordinate homoleptic cerium(III) ions with one ion being disordered (Ce–O distances from 2.487(6) to 2.589(5) Å) around the capsule that is involved in secondary coordination with sulfonate groups (Ce–O⋯O–S distances from 2.606(1) to 2.916(8) Å). Such an intricate arrangement generates narrow channels (approx. diameter = 7 Å) filled with disordered cerium(III) ions and water molecules (Fig. 5). Large tetraphenylphosphonium cations fill the interstices between the capsules in their common phenyl embrace motif like in most reported structures involving the related components.25 Typical interactions involving tetraphenylphosphonium cations and calixarene include C–H⋯π(phenyl) interaction with a C⋯π(phenyl) distance of 3.760 Å and hydrogen bonding with calixarene sulfonate and hydroxyl groups with C–H⋯O of 2.39 to 2.96 Å and 2.59 Å, respectively (corresponding C⋯O distances are 3.28 to 3.39 Å and 3.46 Å, respectively) as the integral part of the hydrogen bonding motifs formed.
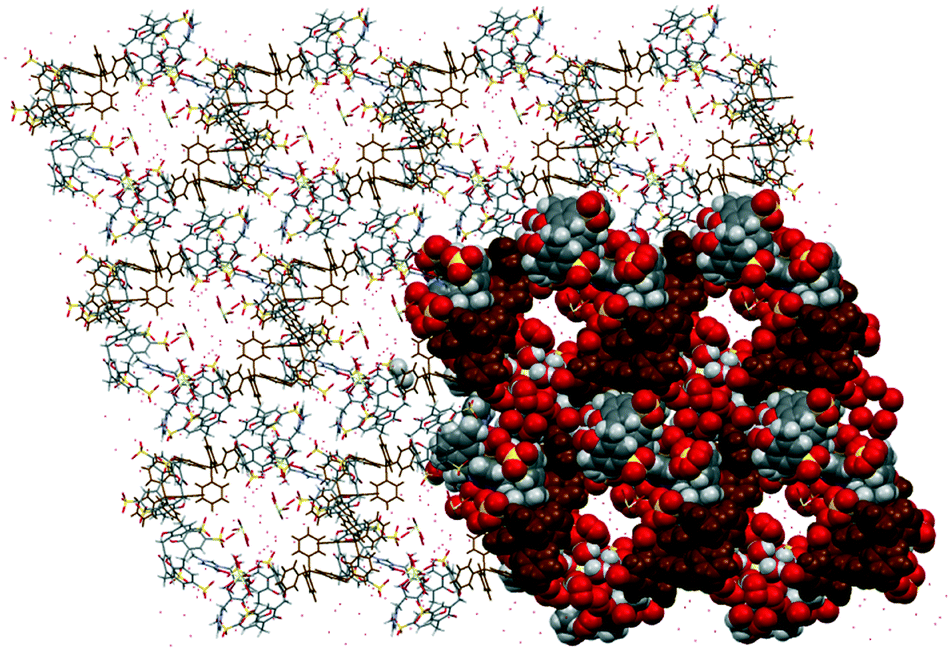 | ||
| Fig. 5 Partial space-filling showing the overall assembly of complex VIII (projection along the a-axis). Hydrophilic channels are evident with water molecules and cerium(III) ions filling the narrow channels. | ||
The extended structure of complex IX exhibits another example that departs from the bilayer arrangement (Fig. 6). The intricate assembly is composed of alternate layers of skewed molecular capsules that are arranged diagonally in opposite directions. Each capsule with a guest molecule fits snugly in the internal space and is coordinated by heteroleptic gadolinium(III) ions (Gd–OS from 2.357(5) to 2.381(6) Å) at the sulfonate groups and sodium ions (Na–O from 2.645(8) to 2.861(8) Å) at both sulfonate and phenolic groups, forming polymeric chains of calixarenes as part of the capsule configuration along with π(phenyl)⋯π(phenyl) stacking of two calixarenes contributing to the polymeric network, with a π(phenyl)⋯π(phenyl) distance of 3.50 Å. Tetraphenylphosphonium molecules similarly fill the interstices of the extended structure but not in the common phenyl embrace manner. These molecules are involved in extensive hydrogen bonding with calixarene sulfonates, phenolics and methylene bridges with C–H⋯O and C–H⋯H–C distances ranging from 2.73 to 2.75 Å and 2.67 to 2.73 Å, respectively.
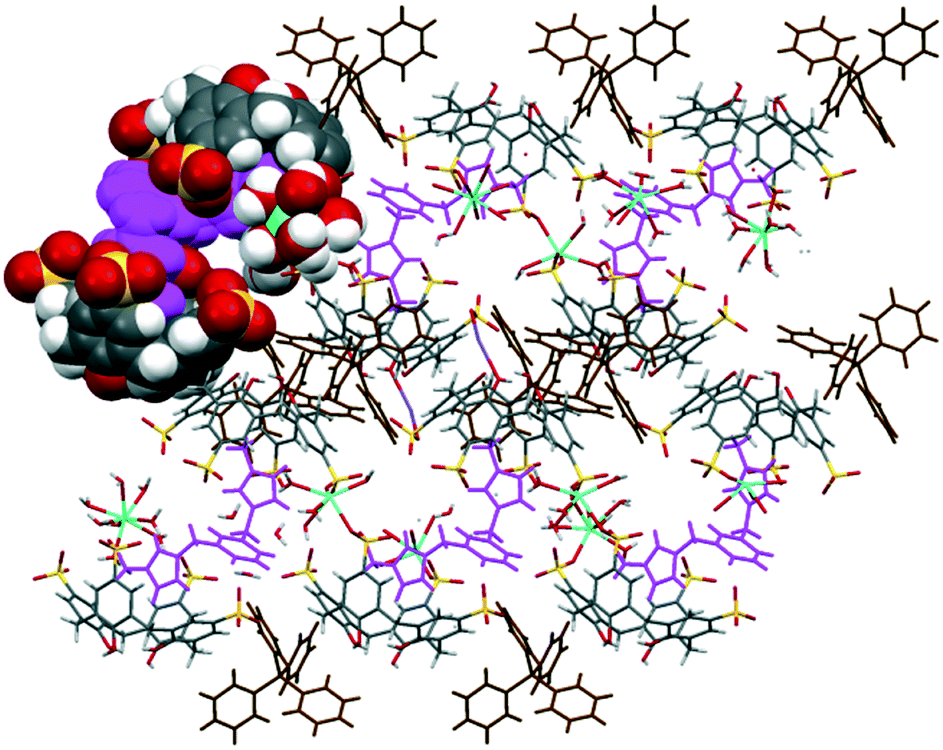 | ||
| Fig. 6 Overall assembly of complex IX showing a layer of skewed molecular capsules (partial space-filled) that are arranged diagonally. | ||
Solution studies
The interaction of 2, 3 and 4 with 1 in D2O at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 mixtures of two species was investigated by using 1H NMR spectroscopy at 25 °C (Fig. 7–9). The interplay of 2 and 1 showed the formation of a host–guest complex in solution, with the largest upfield shift experienced by the protons of methylimidazolium, which demonstrates the shielding effect as a result of calixarenes interacting with the five-membered ring. At the 1:1 ratio of both samples, the overall upfield shifts decreased presumably due to incomplete or partial inclusion. All the protons in 3 experienced an upfield shift, with the n-butyl chain protons showing the largest shift amongst other protons. Reduced upfield shifts are evident for the 1:1 ratio of 3 and 1, similarly, predicting incomplete host–guest inclusion. The interplay of 4 and 1 in solution indicates that the methyl and ring protons have the largest shift amongst other protons and this confirms that the imidazolium moiety is taken up into the calixarene cavity. The presence of additional minor peaks is associated with the residual solvent from dication syntheses.
:1 and 1:2 mixtures of two species was investigated by using 1H NMR spectroscopy at 25 °C (Fig. 7–9). The interplay of 2 and 1 showed the formation of a host–guest complex in solution, with the largest upfield shift experienced by the protons of methylimidazolium, which demonstrates the shielding effect as a result of calixarenes interacting with the five-membered ring. At the 1:1 ratio of both samples, the overall upfield shifts decreased presumably due to incomplete or partial inclusion. All the protons in 3 experienced an upfield shift, with the n-butyl chain protons showing the largest shift amongst other protons. Reduced upfield shifts are evident for the 1:1 ratio of 3 and 1, similarly, predicting incomplete host–guest inclusion. The interplay of 4 and 1 in solution indicates that the methyl and ring protons have the largest shift amongst other protons and this confirms that the imidazolium moiety is taken up into the calixarene cavity. The presence of additional minor peaks is associated with the residual solvent from dication syntheses.
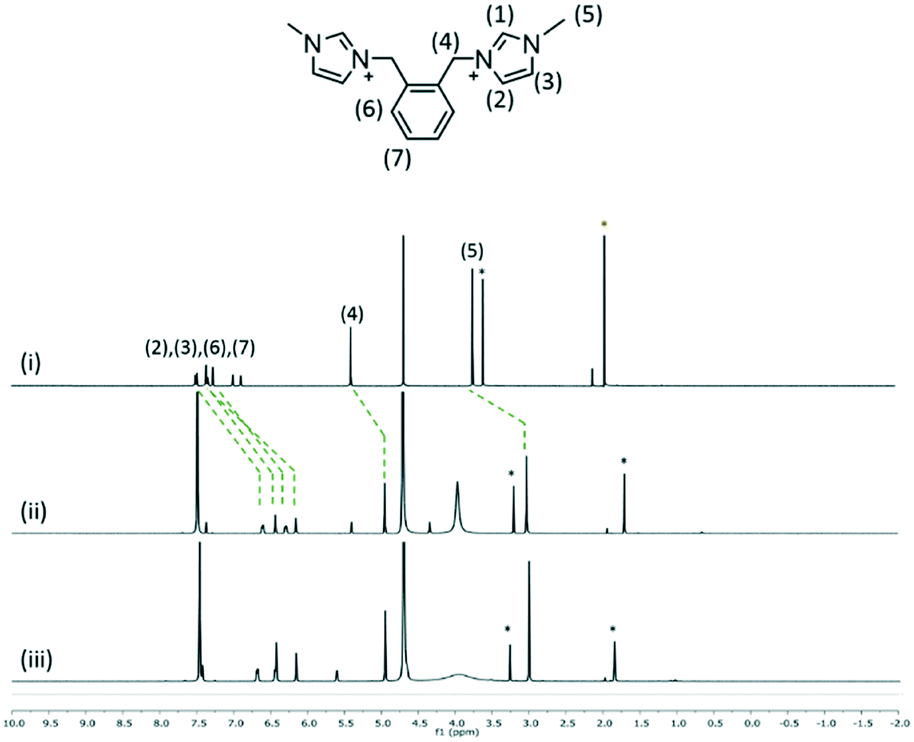 | ||
| Fig. 7
1H NMR spectra of (i) 2 and the interplay of 1 and 2 at the 2:1 ratio (ii) and 1:1 ratio (iii). | ||
 | ||
| Fig. 8
1H NMR spectra of (i) 3 and the interplay of 1 and 3 at the 2:1 ratio (ii) and 1:1 ratio (iii). | ||
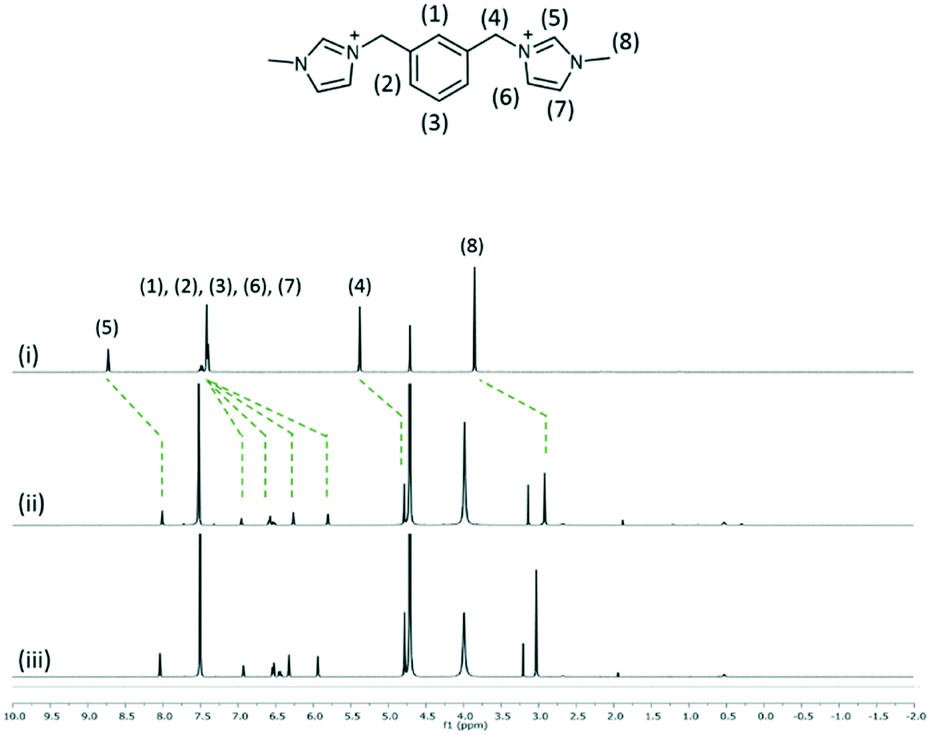 | ||
| Fig. 9
1H NMR spectra of (i) 4 and the interplay of 1 and 4 at the 2:1 ratio (ii) and 1:1 ratio (iii). | ||
Conclusion
In the present study, we have established the self-assembly of multi-component complexes involving N-alkylimidazolium dications possessing different N-heterocyclic substituents substituted at different positions on a bis(methylene)phenyl ring in the presence of water-soluble p-sulfonatocalix[4]arene, monophosphonium cations, and various lanthanide(III) cations. The ability to prepare complexes based on the assembly of three different dications is noteworthy, as the host–guest interplay of the species in the structures depends on the nature of the substituent position on the bis(methylene)phenyl ring (ortho- versus meta-) and the length of terminal alkyl chains (methyl versus butyl).A feature of all the complexes involving either the ortho-disubstituted alkylimidazolium or the meta-disubstituted alkylimidazolium is that the calixarene cavity is occupied by either the bis(methylene)phenyl ring or one of the termini of the dication forming a molecular capsule (or skewed molecular capsule). Such inclusion behaviour is rather different when the para-disubstituted alkylimidazolium is included in the p-sulfonatocalix[4]arene system where the inclusion of the bis(methylene)phenyl ring was never observed. Most of the complexes are self-assembled into a bilayer array except for complexes I, VIII and IX and the interplay of the lanthanides and phosphonium cations is also observed to influence and control the self-assembly of calixarenes. This finding ascertains a new direction to prepare multi-component systems with higher complexities along with the adaptation of different cations, including varying the calixarene sizes and the choice of lanthanide ions, and is set to introduce more diversified functional materials using these dications.
Experimental
Synthesis of ortho- and meta-disubstituted dications
2, 3 and 4 were synthesized with slight modifications according to the literature procedures.223,3′-(1,2-Phenylenebis(methylene))bis(1-methyl-1H-imidazol-3-ium), 2
α,α′-Dibromo-o-xylene (2 mmol) was dissolved in dried acetonitrile (20 mL) and stirred, followed by dropwise addition of 1-methylimidazole (4 mmol). The mixture was stirred for 48 hours at room temperature to give the respective quaternized salts. The solvent was removed using a rotary evaporator and the residue was washed with acetone to remove the unreacted imidazole. The solvent was removed again using the rotary evaporator to afford pure compounds. Yield 93%; white powder; 1H NMR (400 MHz, D2O) ∂H 7.6 (s, 2H; imid-H), 7.4 (m, 4H; Ar–H), 7.3 (s, 2H; imid-H); 5.4 (s, 4H; Ar–CH2), 3.7 (s, 6H; imid-CH2); 13C NMR (100 MHz, D2O) ∂C 131.51, 131.06, 130.67, 123.87, 122.26, 49.97, 35.81.3,3′-(1,2-Phenylenebis(methylene))bis(1-butyl-1H-imidazol-3-ium) dibromide, 3
α,α′-Dibromo-o-xylene (2 mmol) was dissolved in dried acetonitrile (20 mL) and stirred, followed by dropwise addition of 1-butylimidazole (4 mmol). The mixture was stirred for 48 hours at room temperature to give the respective quaternized salts. The solvent was removed using a rotary evaporator and the residue washed with acetone to remove the unreacted imidazole. The solvent was removed again using the rotary evaporator to afford pure compounds. Yield 88%; white powder; 1H NMR (400 MHz, D2O) ∂H 7.4 (m, 8H; Ar–H, imid-H), 5.4 (s, 4H; Ar–CH2), 4.1 (m, 4H; imid-CH2), 1.7 (m, 4H; imid-CH2CH2), 1.3 (m, 4H; imid-CH2CH2CH2), 0.8 (t, 6H; imid-CH2CH2CH2CH3); 13C NMR (100 MHz, D2O) ∂C 131.54, 131.15, 130.73, 122.74, 122.35, 50, 49.57, 31.25, 18.79, 12.59.3,3′-(1,3-Phenylenebis(methylene))bis(1-methyl-1H-imidazol-3-ium) dibromide, 4
α,α′-Dibromo-m-xylene (2 mmol) was dissolved in dried acetonitrile (20 mL) and stirred, followed by dropwise addition of 1-methylimidazole (4 mmol). The mixture was stirred for 48 hours at room temperature to give the respective quaternized salts. The solvent was removed using a rotary evaporator and the residue washed with acetone to remove the unreacted imidazole. The solvent was removed again using the rotary evaporator to afford pure compounds. Yield 90%; white powder; 1H NMR (400 MHz, D2O) ∂H 7.6 (s, 2H; imid-H), 7.4 (m, 4H; Ar–H), 7.3 (s, 2H; imid-H), 5.4 (s, 4H; Ar–CH2), 3.7 (s, 6H; imid-CH3); 13C NMR (100 MHz, D2O) ∂C 131.51, 131.06, 130.67, 123.87, 122.26, 49.97, 35.81.Synthesis of complexes
1, PPh4+Br, CeCl3·7H2O, GdCl3·6H2O and ErCl3·6H2O were purchased from Sigma Aldrich and used as received. A hot solution of CeCl3·7H2O, ErCl3·6H2O or GdCl3·6H2O (1.5 equiv.) was added to a hot solution containing 2, 3 or 4 (1 equiv.) and 1 (2 equiv.) with or without tetraphenylphosphonium bromide (1 equiv.) in a mixture of water and THF (2:1, 1.5 mL). The prepared solution was left to cool and evaporate slowly, with suitable crystals (yield = 50–70%) forming after several days.
Crystallography
All the data were measured from single crystals using an Oxford Diffraction Xcalibur or Gemini-R Ultra CCD diffractometer at T = 100(2) K with monochromatic MoKα (λ = 0.71073 Å) and CuKα (λ = 1.54178 Å) radiation. The data were corrected for Lorentz and polarization effects, and absorption correction was applied using multiple symmetry equivalent reflections. The structures were solved by direct methods and refined against F2 with full-matrix least-squares using the program suite SHELX-2014.26 Anisotropic displacement parameters were employed for the non-hydrogen atoms. All hydrogen atoms were added at calculated positions and refined by the use of a riding model with isotropic displacement parameters based on those of the parent atom. The crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre.Crystal/refinement details for complex I
C112H120Gd2Na2O84S168−, C56H69.333Gd2O46.667S82−, 2(C28H20O16S44−), H28Gd2O146+, 6(C16H20N42+), 41.333(H2O), C320H460Gd6N24Na2O218S32, M = 10146.49, colourless plates, 0.241 × 0.172 × 0.099 mm3, triclinic, space group P (no. 2), a = 17.6570(5), b = 21.7188(5), c = 27.9006(5) Å, α = 97.688(2), β = 100.408(2), γ = 98.551(2)°, V = 10263.6(4) Å3, Z = 1, Dc = 1.642 g cm−3, F000 = 5210, 2θmax = 134.7°, 100035 reflections collected, 36345 unique (Rint = 0.0616). Final GooF = 1.401, R1 = 0.1047, wR2 = 0.2410, R indices based on 28463 reflections with I > 2σ(I), |Δρ|max = 7.2(2) e Å−3, 2934 parameters, 410 restraints. CCDC number 1951748.
Crystal/refinement details for complex II
2(C28H20O16S44−), C22H32N42+, 2(CeH18O93+),Cl−, Na+, 13(H2O), C78H134Ce2ClN4NaO63S8, M = 2731.04, colourless plates, 0.358 × 0.331 × 0.077 mm3, triclinic, space group P (no. 2), a = 13.8053(3), b = 13.8709(3), c = 16.1942(4) Å, α = 65.439(2), β = 83.047(2), γ = 87.191(2)°, V = 2799.73(12) Å3, Z = 1, Dc = 1.620 g cm−3, μ = 1.084 mm−1. F000 = 1406, 2θmax = 64.9°, 46757 reflections collected, 46757 unique. Final GooF = 1.007, R1 = 0.0434, wR2 = 0.1076, R indices based on 40056 reflections with I > 2σ(I), |Δρ|max = 1.3(1) e Å−3, 800 parameters, 62 restraints. CCDC number 1951749.
Crystal/refinement details for complex III
2(C28H34ErO23S4−), 2(C28H21O16S43−), 4(C22H32N42+), Cl−, Na+, 24(H2O), C200H286ClEr2N16NaO102S16, M = 5452.35, colourless prisms, 0.411 × 0.245 × 0.158 mm3, triclinic, space group P (no. 2), a = 16.9848(3), b = 17.2326(3), c = 23.7694(4) Å, α = 98.350(1), β = 103.459(1), γ = 116.030(2)°, V = 5831.1(2) Å3, Z = 1, Dc = 1.553 g cm−3, μ = 0.974 mm−1. F000 = 2834, 2θmax = 65.5°, 126710 reflections collected, 39660 unique (Rint = 0.0438). Final GooF = 1.070, R1 = 0.0530, wR2 = 0.1289, R indices based on 33615 reflections with I > 2σ(I), |Δρ|max = 4.1(1) e Å−3, 1714 parameters, 107 restraints. CCDC number 1951750.
Crystal/refinement details for complex IV
C28H46CeNaO28S42+, C28H38CeO24S4+, 3(C16H20N42+), 2(C28H20O16S44−), Cl−, 20(H2O), C160H224Ce2ClN12NaO104S16, M = 4831.14, colourless needles, 0.364 × 0.076 × 0.029 mm3, triclinic, space group P (no. 2), a = 13.9722(3), b = 17.1363(7), c = 44.1982(12) Å, α = 97.328(3), β = 93.974(2), γ = 100.205(3)°, V = 10284.1(6) Å3, Z = 2, Dc = 1.560 g cm−3, μ = 5.961 mm−1. F000 = 5000, 2θmax = 136.1°, 87124 reflections collected, 36681 unique (Rint = 0.0988). Final GooF = 1.246, R1 = 0.1084, wR2 = 0.2528, R indices based on 21259 reflections with I > 2σ(I), |Δρ|max = 2.5(2) e Å−3, 2669 parameters, 407 restraints. CCDC number 1951751.
Crystal/refinement details for complex V
2(C28H20O16S44−), 2(C28H25.33Ce0.33O18.67S43−), 2(C16H20N42+), 3.33(CeH18O93+), 33.67(H2O), C144H258Ce4N8O133S16, M = 5303.01, colourless prisms, 0.279 × 0.237 × 0.091 mm3, triclinic, space group P (no. 2), a = 14.9999(3), b = 17.5274(4), c = 21.1206(4) Å, α = 100.434(2), β = 96.481(2), γ = 98.621(2)°, V = 5343.0(2) Å3, Z = 1, Dc = 1.648 g cm−3, μ = 1.108 mm−1. F000 = 2730, 2θmax = 65.2°, 116456 reflections collected, 36073 unique (Rint = 0.0659). Final GooF = 1.005, R1 = 0.1066, wR2 = 0.2585, R indices based on 23683 reflections with I > 2σ(I), |Δρ|max = 4.5(2) e Å−3, 1540 parameters, 168 restraints. CCDC number 1951752.
Crystal/refinement details for complex VI
2(C28H20O16S44−), C16H20N42+, GdH16O83+, GdH18O93+, 16(H2O), C72H126Gd2N4O65S8, M = 2658.74, colourless plates, 0.346 × 0.326 × 0.117 mm3, triclinic, space group P (no. 2), a = 15.2241(2), b = 17.7001(3), c = 20.5479(3) Å, α = 100.785(1), β = 96.463(1), γ = 97.675(1)°, V = 5336.39(14) Å3, Z = 2, Dc = 1.655 g cm−3, μ = 1.497 mm−1. F000 = 2724, 2θmax = 64.8°, 113283 reflections collected, 35163 unique (Rint = 0.0365). Final GooF = 1.000, R1 = 0.0354, wR2 = 0.0813, R indices based on 29338 reflections with I > 2σ(I), |Δρ|max = 1.4(1) e Å−3, 1568 parameters, 116 restraints. CCDC number 1951753.
Crystal/refinement details for complex VII
2(C28H20O16S44−), C16H20N42+, [0.5(C4H22ErO83+)/0.5(H16ErO83+)], [0.5(H16ErO83+)/0.5(H14ErO73+)], 16(H2O), C74H126Er2N4O63.50S8, M = 2678.78, colourless plates, 0.493 × 0.359 × 0.175 mm3, triclinic, space group P (no. 2), a = 15.0362(2), b = 17.4614(3), c = 21.0544(3) Å, α = 100.755(1), β = 95.900(1), γ = 98.544(1)°, V = 5321.84(14) Å3, Z = 2, Dc = 1.672 g cm−3, μ = 1.831 mm−1. F000 = 2740, 2θmax = 61.0°, 111116 reflections collected, 32474 unique (Rint = 0.0323). Final GooF = 1.003, R1 = 0.0752, wR2 = 0.1866, R indices based on 27537 reflections with I > 2σ(I), |Δρ|max = 4.2(72) e Å−3, 1569 parameters, 265 restraints CCDC number 1951754.
Crystal/refinement details for complex VIII
2(C28H20O16S44−), 2(C24H20P+), C16H20N42+, (CeH18O93+), 0.33(Ce3+), 22(H2O), C120H162Ce1.33N4O63P2S8, M = 3174.01, colourless needles, 0.365 × 0.145 × 0.053 mm3, triclinic, space group P (no. 2), a = 15.1413(3), b = 21.5216(4), c = 25.0716(4) Å, α = 105.589(1), β = 96.342(1), γ = 109.239(2)°, V = 7249.8(2) Å3, Z = 2, Dc = 1.454 g cm−3, μ = 5.315 mm−1. F000 = 3299, 2θmax = 134.7°, 171641 reflections collected, 25781 unique (Rint = 0.0711). Final GooF = 1.015, R1 = 0.0855, wR2 = 0.2439, R indices based on 20437 reflections with I > 2σ(I), |Δρ|max = 2.0(1) e Å−3, 1985 parameters, 172 restraints. CCDC number 1951755.
Crystal/refinement details for complex IX
C576H812ClGd10N24NaO358P6S48, M = 17156.09, colourless plates, 0.373 × 0.246 × 0.129 mm3, monoclinic, space group P21/c (no. 14), a = 22.4311(3), b = 36.5505(4), c = 43.3752(8) Å, β = 104.433(2)°, V = 34439.6(9) Å3, Z = 2, Dc = 1.654 g cm−3, μ = 8.621 mm−1. F000 = 17652, 2θmax = 135.7°, 285014 reflections collected, 61897 unique (Rint = 0.0675). Final GooF = 1.008, R1 = 0.0975, wR2 = 0.2571, R indices based on 45935 reflections with I > 2σ(I), |Δρ|max = 4.9(2) e Å−3, 4969 parameters, 683 restraints. CCDC number 1951756.
Solution studies
All the 1H NMR spectra were collected on a Bruker Avance III 400 MHz spectrometer using D2O at 25 °C. All the chemical shift changes for all the proton signals of 2, 3 and 4 in the presence of 1 were monitored and analysed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Monash University Malaysia, the University of Western Australia and the University of Malaya for supporting this work. This work is also supported by the Fundamental Research Grant Scheme, Ministry of Education Malaysia (FRGS/1/2018/STG07/MUSM/03/1).References
- R. Delgado, Rev. Port. Quim., 1995, 2, 18 CAS.
- V. Alexander, Chem. Rev., 1995, 95, 273 CrossRef CAS.
- S. J. Swamy and S. Pola, Spectrochim. Acta, Part A, 2008, 70, 929 CrossRef CAS.
- S. Chandra, D. Jain, A. K. Sharma and P. Sharma, Molecules, 2009, 14, 174 CrossRef CAS PubMed.
- S. J. Archibald, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2009, 105, 297 RSC.
- S. Patra, D. Maity, R. Gunupuru, P. Agnihotri and P. Paul, J. Chem. Sci., 2012, 124, 1287 CrossRef CAS.
- D. S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925 CrossRef CAS PubMed.
- F. Perret, A. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425 RSC.
- R. Ludwig, Microchim. Acta, 2005, 152, 1–19 CrossRef CAS.
- N. Douteau-Guével, F. Perret, A. W. Coleman, J.-P. Morel and N. Morel-Desrosiers, J. Chem. Soc., Perkin Trans. 2, 2002, 524 RSC.
- N. Basilio, V. Francisco and L. Garcia-Rio, Int. J. Mol. Sci., 2013, 14, 3140 CrossRef CAS.
- J. L. Atwood, G. W. Orr, F. Hamada, R. L. Vincent, S. G. Bott and K. D. Robinson, J. Inclusion Phenom. Macrocyclic Chem., 1992, 14, 37 CrossRef CAS.
- J. L. Atwood, G. W. Orr and K. D. Robinson, Supramol. Chem., 1994, 3, 89 CrossRef CAS.
- J. L. Atwood, G. W. Orr, F. Hamada, R. L. Vincent, S. G. Bott and K. D. Robinson, J. Am. Chem. Soc., 1991, 113, 2760 CrossRef CAS.
- B. T. Zhao, H. Wang, H. Y. Zhang and Y. Liu, J. Mol. Struct., 2005, 740, 101 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, M. J. Hardie and C. L. Raston, Coord. Chem. Rev., 2001, 222, 3 CrossRef CAS.
- I. Ling, Y. Alias, A. N. Sobolev and C. L. Raston, CrystEngComm, 2010, 12, 573 RSC.
- I. Ling, Y. Alias, A. N. Sobolev, L. T. Byrne and C. L. Raston, Chem. – Eur. J., 2010, 16, 6973 CrossRef CAS.
- I. Ling, A. N. Sobolev, Y. Alias and C. L. Raston, CrystEngComm, 2013, 15, 2888 RSC.
- I. Ling, Y. Alias, A. N. Sobolev, L. T. Byrne and C. L. Raston, CrystEngComm, 2011, 13, 787 RSC.
- I. Ling, Y. Alias, M. S. A. Rahim, B. W. Skelton, L. T. Byrne and C. L. Raston, Aust. J. Chem., 2012, 65, 755 CrossRef CAS.
- I. Ling, A. N. Sobolev, K. Sabah and R. Hashim, CrystEngComm, 2015, 17, 5841 RSC.
- I. Ling, A. N. Sobolev and C. L. Raston, CrystEngComm, 2016, 18, 4929 RSC.
- I. Ling, Y. Alias, A. N. Sobolev and C. L. Raston, CrystEngComm, 2010, 12, 1869 RSC.
- I. Ling, Y. Alias and C. L. Raston, New J. Chem., 2010, 34, 1802 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1951748–1951756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01410a |
| This journal is © The Royal Society of Chemistry 2020 |