
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient method to prepare supported bismuth nanoparticles as highly selective electrocatalyst for the conversion of CO2 into formate†
Matteo
Miola
 ,
Bart C. A.
de Jong
and
Paolo P.
Pescarmona
*
,
Bart C. A.
de Jong
and
Paolo P.
Pescarmona
*
Chemical Engineering Group, Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
First published on 3rd November 2020
Abstract
We report a cost-effective, straightforward synthesis of a novel electrocatalyst for the reduction of CO2 to formate, which achieves nearly complete Faradaic efficiency (FE) at an overpotential (η) of 0.88 V under ambient conditions. The electrocatalyst was prepared using bismuth subsalicylate as precursor and consists of bismuth nanoparticles (Bi NPs) with an average diameter of 5.5 nm supported on activated carbon.
Anthropogenic CO2 emissions have been identified as a major factor impacting environment and climate.1,2 Therefore, it is of paramount importance to decrease the atmospheric CO2 concentration through its capture and conversion into valuable chemicals and fuels. One approach to achieve this is through the electrochemical reduction of CO2 to C1 products.3 The electrochemical approach is particularly appealing because it can use intermittent renewable power sources (e.g. wind and solar) and it can be performed at room temperature and pressure. Among the various products of the electrochemical conversion of CO2 into C1 products (e.g. CO, HCOO−, CH4),4 formate is a relevant target owing to its relatively high accessibility (the reduction of CO2 to HCOO− requires only 2 e−) and to its broad range of applications, e.g. in the leather and textile manufacturing,5 as preserving and antibacterial agent,6 as fuel in fuel cells7 and as additive in cement production.8
In a basic aqueous medium such as 0.5 M NaHCO3 (pH 8.4) or CO2-saturated 0.5 M NaHCO3 (pH 7.3), the electrochemical reduction of CO2 towards formate in the cathodic cell compartment is described by eqn (1), and it is coupled with the oxygen evolution reaction in the anodic compartment (eqn (2)).
| CO2 + 2e− + H2O → HCO2− + OH− | (1) |
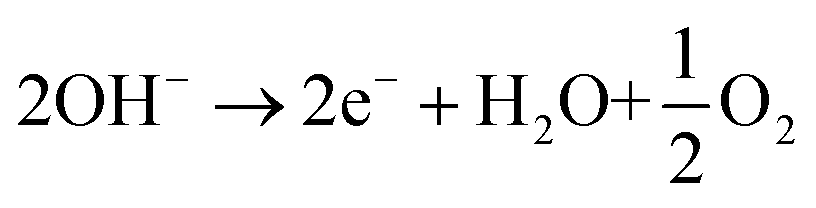 | (2) |
The development of a suitable electrocatalyst is crucial in order to achieve the electrochemical reduction of CO2 to the chosen product with high activity (current density) and selectivity (Faradaic efficiency). The state-of-the-art electrocatalysts for the conversion of CO2 into formate are typically based on main group metals such as Sn, Pb, Bi and In.9 It has been proposed that these metals promote the adsorption of CO2 with subsequent formation of the *OCOH over the *COOH intermediate on the catalyst surface. The *OCOH intermediate is generally recognised as the precursor to the formation of formate as product of CO2 reduction.9 Experimental evidences suggest that the oxide layer that tends to form at the surface of these main group metals has a central role in defining their catalytic properties.9 Among these metals, Bi is particularly appealing for the low cost, low toxicity and low environmental hazard. Bi-based electrocatalysts have been prepared through the reduction of Bi3+ precursors via chemical,10–14 electrochemical15–23 or hydrothermal24–27 methods (Tables S1, S2 and Fig. S1, ESI†). In few cases, the reduction step was combined with oxidative calcination treatments to tune the features of the surface oxide layer.10,21,28 These synthesis procedures require costly multiple steps and/or hazardous reactants (Table S2, ESI†), thus posing limitations to the industrial upscaling of these electrocatalysts.
In this communication, we report a straightforward, cost-effective and fast synthesis method to produce a highly selective and active Bi-based electrocatalyst for the reduction of CO2 to formate. This novel synthesis method was designed to exploit the controlled, thermal reduction of an organic bismuth precursor on activated carbon (AC) in inert atmosphere (i.e. pyrolysis) to generate well-dispersed Bi nanoparticles. The synthesis consists of three consecutive steps involving wet impregnation of the activated carbon support with a Bi precursor, drying and pyrolysis (Fig. 1). After this procedure, the electrocatalyst can be readily employed without requiring further purification or activation steps. We reasoned that bismuth subsalicylate (BiSub) would be an ideal Bi precursor for the aforementioned approach, owing to the bidentate coordination of the subsalicylate to the Bi atom, the aromatic backbone and the delocalised electronic structure that extends over the whole ligand. The combination of these features is expected to lead to strong physisorption on the activated carbon support, thus hindering the mobility of the BiSub precursor during the thermal treatment. In turn, this is anticipated to favour nucleation over growth, leading to the formation of small Bi nanoparticles. Indeed, the electrocatalyst produced by pyrolysis at 400 °C of the material obtained by supporting BiSub on activated carbon (BiSub@AC-400), consisted of small and highly dispersed Bi nanoparticles (Bi NP, 5.5 ± 2 nm) on AC, as proven by TEM analysis (Fig. 2 and Fig. S2, S3, ESI†). The high control of the particle size and distribution achieved with our method is notable, particularly when considering that the synthesis methods reported in the literature to prepare Bi-based electrocatalysts either require complex and expensive procedures to produce nanoparticles12,13,26 or generate materials with micrometric scale at least in one dimension (such as in the case of nanoflakes,17,21,29 nanosheets,16,18,19,22,25,27,28 clusters,11,30 dendrites,20 needles,15,24 or tubes10). The loading of Bi in the BiSub@AC-400 electrocatalyst was estimated to be 47 wt% based on ICP-OES analysis and supported by TGA characterisation (Tables S3, S4 and Fig. S4a, ESI†). The specific surface area of BiSub@AC-400 is rather high (358 m2 g−1, see Fig. 3a), particularly when taking into account the high loading of bismuth in this material. Upon the adsorption of BiSub on AC and pyrolysis, the specific surface area decreased of 54% compared to the AC support alone (Fig. 3a). The same decrease was observed for the electrochemical surface area (ECSA) determined by capacitance measurements (Fig. S5, ESI†). Notably, the pyrolysis at 400 °C did not affect the surface area, as shown by the analogous values obtained prior to and after the thermal treatment (Fig. 3a). This is attributed to the structural stability of the activated carbon support under the employed pyrolysis conditions, which was demonstrated by TGA analysis under N2 atmosphere (Fig. 3b). TGA also allowed monitoring the effect of the pyrolysis treatment on the bismuth precursor during the synthesis of the supported Bi nanoparticles. Whereas the degradation of the pure BiSub precursor occurred in the 240–400 °C interval, in the presence of the activated carbon support the degradation covered a larger temperature range (Fig. 3b and Fig. S4b, ESI†), in line with the anticipated strong interaction between BiSub and the support. XRD characterisation of BiSub@AC-400 (Fig. 3c) showed that the nanoparticles consist of three main crystalline phases: bismutite (Bi2CO3O2), bismuth oxide (Bi2O3) and metallic bismuth (Bi0). The nanoparticles are characterised by a metallic core, as indicated by the dspacing = 0.32 ± 0.02 nm assigned to the (012) Bi0 planes,26 which was obtained from HR-TEM analysis (Fig. 2b). A sub-nanometric layer containing oxygen could be observed at the surface of some nanoparticles (Fig. 2b and d), in accordance with the presence of the Bi2CO3O2 and Bi2O3 phases identified by XRD. These results demonstrate that the pyrolysis treatment at 400 °C promotes the reduction of the Bi3+ present in the bismuth subsalicylate precursor to Bi0. Altogether, the characterisation study proved that BiSub@AC-400 has a number of promising features for application as electrocatalyst: a relatively high specific surface area and a high loading of Bi organised in small, highly dispersed nanoparticles consisting of a metallic Bi core surrounded by a layer of oxidised Bi species, on a conductive support. The electrocatalytic performance of BiSub@AC-400 in the electrochemical reduction of carbon dioxide was studied in an H-cell configuration (Fig. S6, ESI†), employing a CO2-saturated 0.5 M NaHCO3 electrolyte. The cathodic half-cell potential was applied in the range of −0.87 to −1.37 V vs. RHE and kept at each potential for 30 min, to measure the current density (j) and Faradaic efficiency (FE), Fig. 4a. The electrocatalyst was able to promote the reduction of CO2 resulting in the highly selective production of formate, with minor amounts of H2 in the gas phase as the only side-product (Fig. S7, ESI†). The Faradaic efficiency towards the desired formate product was 83% at an overpotential (η) as low as 0.68 V (i.e. at E = −0.87 V vs. RHE). A nearly complete selectivity (FE > 99%) towards formate was achieved at E = −1.07 V vs. RHE, corresponding to a η = 0.88 V with a current density of 6.2 mA cm−2. The current density steadily increased at higher potentials up to j = 11.3 mA cm−2 at E = −1.37 V vs. RHE, although this was coupled to a decrease in Faradaic efficiency (Fig. 4b), caused by the competing hydrogen evolution reaction. The Tafel plot analysis (Fig. S8, ESI†) revealed a slope of 186 mV dec−1, which is much larger than the value of 118 mV dec−1 that would be expected if the first electron transfer (CO2 + e− → CO2˙−) were the rate-determining step. This suggests that the second electron transfer and the concomitant protonation of the adsorbed CO2 radical anion are the rate-determining step over BiSub@AC-400.31
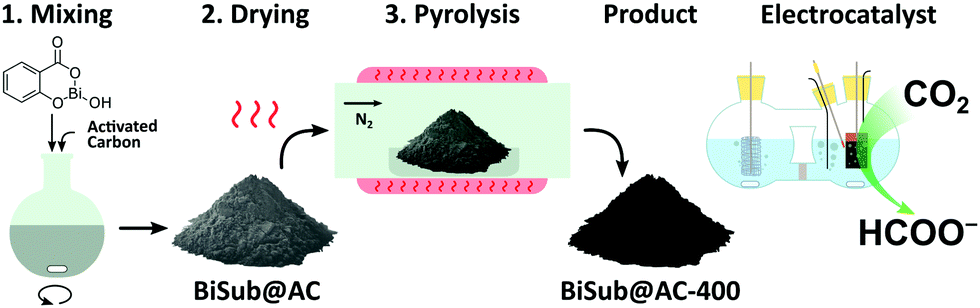 | ||
| Fig. 1 Synthesis scheme of the BiSub@AC-400 electrocatalyst. | ||
 | ||
| Fig. 2 (a) HR-TEM pictures of BiSub@AC-400: with (b) focus on the crystal lattice and the surface of the nanoparticles; (c) HAADF-STEM and (d) EDX-STM pictures. | ||
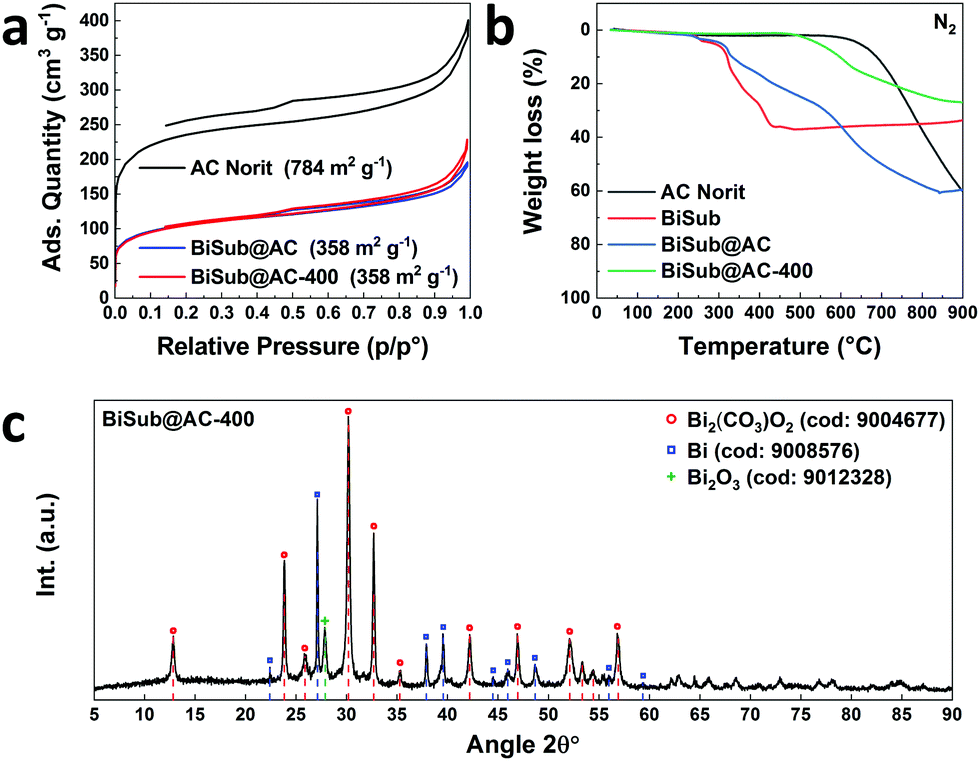 | ||
| Fig. 3 (a) N2 physisorption analysis of AC, BiSub@AC and BiSub@AC-400; (b) TGA analysis under N2 atmosphere of AC, BiSub, BiSub@AC and BiSub@AC-400; (c) XRD characterisation of BiSub@AC-400. | ||
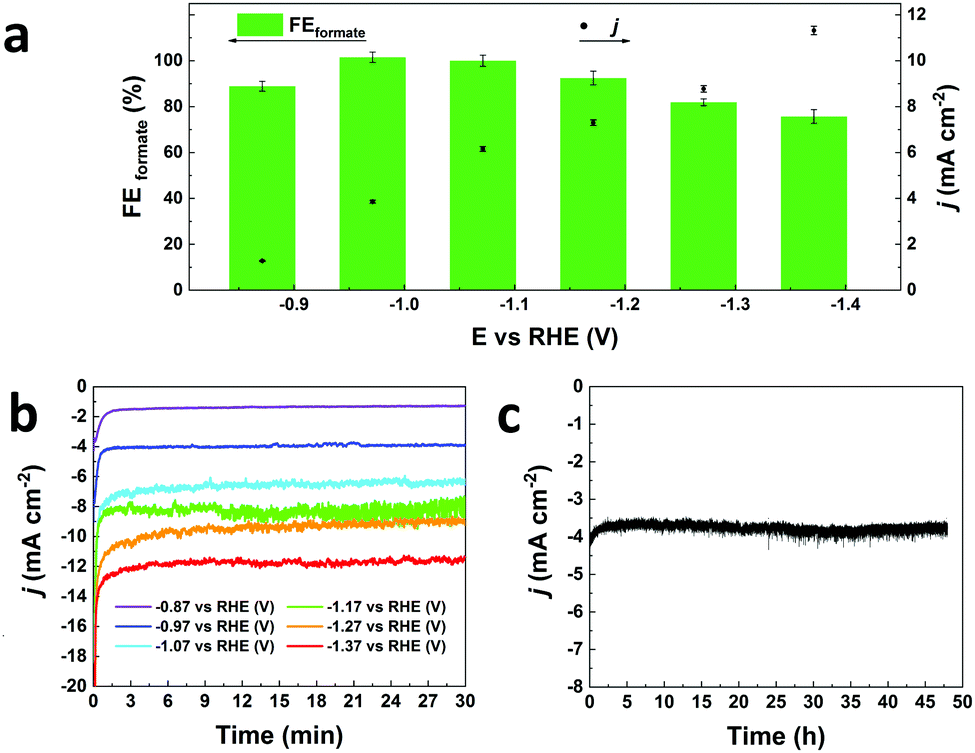 | ||
| Fig. 4 (a) Faradaic efficiency (FE, green bars) and current density (j, black dots) in the reduction of CO2 to formate with BiSub@AC-400 as electrocatalyst in CO2-saturated 0.5 M NaHCO3 at the indicated potentials; (b) the corresponding chronoamperometric study at various potentials; (c) 48 h chronoamperometry at E = −0.97 V vs. RHE. | ||
The electrocatalyst displayed high stability in a chrono-amperometric test at E = −0.97 V vs. RHE, in which it displayed a constant current of j = 3.8 mA cm−2 for 48 h (Fig. 4c). During this stability test, the nearly complete selectivity towards formate is retained in the first 24 h (FE = 99%), after which the selectivity decreases slightly (FE = 90% after 48 h). ICP-OES analysis of the electrolyte solution after the chronoamperometric test indicated that bismuth leaching was absent or below the detection limit (<0.07 mg L−1, i.e. <1% of the Bi content). TEM analysis of the electrode material after the stability test (Fig. S9, ESI†) revealed that the bismuth nanoparticles were preserved, though their average diameter slightly increased to 11 ± 5 nm and some rare particle aggregation was observed (Fig. S9a and S10, ESI†). Yet, several nanoparticles with diameter <6 nm were detected (Fig. S9f, ESI†).
Our synthesis method is of general validity and allowed preparing electrocatalysts also with different Bi loading, namely by employing either half or double the amount of BiSub compared to that used in BiSub@AC-400 (Table S5, ESI†). These preliminary studies on the effect of the Bi content showed that the loading used in the synthesis of BiSub@AC-400 leads to the optimum performance both in terms of current density and Faradaic efficiency towards formate (Fig. S11, ESI†). When the activated carbon (with no bismuth supported on it) was tested as electrocatalyst under the same conditions, a much lower current density was achieved compared to BiSub@AC-400 (Fig. S11, ESI†), and no formate was observed (FE < 1%). This result confirms the anticipated role of the bismuth species in catalysing the electrochemical reduction of CO2 to formate.
Finally, the electrocatalytic performance of BiSub@AC-400 was compared to that of three electrocatalysts prepared via electrodeposition of bismuth at three potentials (−0.67 V, −0.87 V and −1.07 V vs. RHE) on the same activated carbon support and with an analogous total Bi content. The obtained electrocatalysts displayed lower formate selectivity (FE < 86%) and comparable yet slightly lower current density than BiSub@AC-400 (Fig. S12, ESI†). These results highlight the very good electrocatalytic performance of BiSub@AC-400 in the reduction of CO2 to formate. We hypothesise that the lower Faradaic efficiency towards formate observed with the electrocatalysts prepared by electrochemical deposition stems from a larger fraction of surface of activated carbon remaining exposed (compared to BiSub@AC-400), which acts as active area for the competing hydrogen evolution reaction.
In conclusion, we reported a novel, straightforward method to synthesise an electrocatalyst consisting of small and highly dispersed Bi nanoparticles on activated carbon by using a commercially available and affordable bismuth precursor. Importantly, the synthesis lends itself to upscaling, requiring only larger synthesis and pyrolysis vessels to produce the electrocatalyst on a multi-gram scale. The electrocatalyst can reach full selectivity towards the desired formate product (FE > 99%) at η = 0.88 V and displays high stability over 48 h electrolysis. Our synthesis method is an attractive alternative to a benchmark approach as electrodeposition, as proven by the higher Faradaic efficiency towards formate displayed by BiSub@AC-400 compared to counterpart electrocatalysts with an analogous Bi loading on the same AC support, but prepared by electrodeposition. Although a thorough comparison with literature electrocatalysts is made difficult by the use of different experimental parameters and cell configurations, the Faradaic efficiency of our electrocatalyst is in the same range as the state-of-the-art Bi-based electrocatalysts for the electrochemical reduction of CO2 to formate (see Table S1 and Fig. S1, ESI†), with the advantage of a highly accessible, cost-effective and upscalable synthesis method (see Table S2, ESI† for a comparison with the synthesis methods of Bi-electrocatalysts from the literature). In perspective, we foresee the direct application of this synthesis method for the preparation on carbon-based gas diffusion layers (GDL), without requiring the use of a binder (e.g. Nafion) to fix the electrocatalyst particles on the GDL. This feature will allow investigating the performance of this electrocatalyst in flow cells, which is the most suitable cell configuration for the large-scale electrochemical reduction of CO2 to formate.
Future work will exploit the high versatility of our synthesis method to deepen the investigation of the bismuth loading and to study the effect of the nature of the metal precursor, of the support and of the pyrolysis treatment, with the purpose of expanding the scope of applicability of our approach and of optimising the electrocatalytic performance.
This work was realised in the context of the RECODE project (Recycling carbon dioxide in the cement industry to produce added-value additives: a step towards a CO2 circular economy, https://www.recodeh2020.eu), which was funded by the H2020 research and innovation program of the European Union (Grant agreement number 768583). We acknowledge Marcel de Vries and Erwin Wilbers for technical support, Jan Henk Marsman and Léon Rohrbach for support with the HPLC and GC analysis, Hans van der Velde for the ICP-OES analysis and Gert H. ten Brink for the TEM analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- IPCC–Intergovernmental Panel on Climate Change 2014: Synthesis Report, 2015.
- NASA–National Aeronautics and Space Administration, https://climate.nasa.gov/.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef.
- BASF, http://formic-acid.basf.us/p02/USWeb-Internet/formic-acid/en_GB/.
- J. L. Thompson and M. Hinton, Br. Poult. Sci., 1997, 38, 59–65 CrossRef CAS.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- RECODE EU project, https://www.recodeh2020.eu/.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 10, 1902338 CrossRef.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef.
- X. Zhang, T. Lei, Y. Liu and J. Qiao, Appl. Catal., B, 2017, 218, 46–50 CrossRef CAS.
- D. Zhang, Z. Tao, F. Feng, B. He, W. Zhou, J. Sun, J. Xu, Q. Wang and L. Zhao, Electrochim. Acta, 2020, 334, 135563 CrossRef CAS.
- B. Avila-Bolivar, L. Garcia-Cruz, V. Montiel and J. Solla-Gullon, Molecules, 2019, 24, 2032 CrossRef CAS.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- J. H. Koh, D. H. Won, T. Eom, N.-K. Kim, K. D. Jung, H. Kim, Y. J. Hwang and B. K. Min, ACS Catal., 2017, 7, 5071–5077 CrossRef CAS.
- W. Lv, J. Bei, R. Zhang, W. Wang, F. Kong, L. Wang and W. Wang, ACS Omega, 2017, 2, 2561–2567 CrossRef CAS.
- S. Kim, W. J. Dong, S. Gim, W. Sohn, J. Y. Park, C. J. Yoo, H. W. Jang and J.-L. Lee, Nano Energy, 2017, 39, 44–52 CrossRef CAS.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef.
- W. Lv, J. Zhou, J. Bei, R. Zhang, L. Wang, Q. Xu and W. Wang, Appl. Surf. Sci., 2017, 393, 191–196 CrossRef CAS.
- J. Bei, Z. Rui, C. Zhidong, L. Weixin and W. Wei, Int. J. Electrochem. Sci., 2017, 12, 2365–2375 CrossRef CAS.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS.
- J. H. Koh, D. H. Won, T. Eom, N.-K. Kim, K. D. Jung, H. Kim, Y. J. Hwang and B. K. Min, ACS Catal., 2017, 7, 5071–5077 CrossRef CAS.
- Y. Zhang, F. Li, X. Zhang, T. Williams, C. D. Easton, A. M. Bond and J. Zhang, J. Mater. Chem. A, 2018, 6, 4714–4720 RSC.
- Z. Chen, K. Mou, X. Wang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 12790–12794 CrossRef CAS.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 1910408 CrossRef CAS.
- F. L. Meng, Q. Zhang, K. H. Liu and X. B. Zhang, Chem. - Eur. J., 2019, 25, 1–7 CrossRef.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2019, 10, 743–750 CrossRef.
- Y. Zhang, X. Zhang, Y. Ling, F. Li, A. M. Bond and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13283–13287 CrossRef CAS.
- Y. Qiu, J. Du, W. Dong, C. Dai and C. Tao, J. CO2 Util., 2017, 20, 328–335 CrossRef CAS.
- J. H. Zhou, K. Yuan, L. Zhou, Y. Guo, M. Y. Luo, X. Y. Guo, Q. Y. Meng and Y. W. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14197–14201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06818g |
| This journal is © The Royal Society of Chemistry 2020 |