
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic chiral cyclohexanohemicucurbit[12]uril†‡
Kamini A.
Mishra
 a,
Jasper
Adamson
b,
Mario
Öeren
c,
Sandra
Kaabel
ad,
Maria
Fomitšenko
a and
Riina
Aav
*a
a,
Jasper
Adamson
b,
Mario
Öeren
c,
Sandra
Kaabel
ad,
Maria
Fomitšenko
a and
Riina
Aav
*a
aDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia tee 15, 12618 Tallinn, Estonia. E-mail: riina.aav@taltech.ee
bChemical Physics Laboratory, National Institute of Chemical Physics and Biophysics, Akadeemia tee 23, 12618 Tallinn, Estonia
cOptibrium Limited, F5-6 Blenheim House, Denny End Road, Cambridge, CB25 9PB, UK
dDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, H3A 0B8, Montreal, Quebec, Canada
First published on 2nd November 2020
Abstract
NMR spectroscopy and DFT modeling studies of chiral cyclohexanohemicucurbit[12]uril indicate that the macrocycle adopts a concave octagonal shape with two distinct conformational flexibilities in solution. Methylene bridge flipping occurs at temperatures above 265 K, while urea monomers rotate at temperatures above 308 K, resulting in the loss of confined space within the macrocycle.
Dynamic molecules capable of adopting distinct shapes in response to external stimuli have driven the development of molecular machines in supramolecular chemistry.1 Of great importance in this field is the ability to control and direct interactions which can be achieved within the confined spaces of macrocycle cavities. Large macrocycles able to repeatedly undergo significant conformational changes while retaining their molecular integrity have attracted particular interest.2,3
Several macrocycles that differ considerably in conformation from their smaller counterparts have been reported. Cyclodextrins (CDs) containing more than 9 glycose units, referred to as large-ring CDs (LR-CDs),4 can assume a helical folding arrangement, e.g., the 26-membered CD.5–9 LR-CDs with 14 units have been shown to adopt a conformation that resembles a concave polygon.10,11 Large pillar[n]arenes, where n = 8–10, containing up to 10 monomers in their structures, also adopt concave polygon shapes.12 Giant calix[n]arenes with up to 90 phenolic subunits have been described;13 however, calix[16]arene is the largest calix[n]arene with a known conformation. It has the shape of two superimposed Celtic torcs.14 The largest reported cucurbituril (CB) contains 15 monomers.15 CBs with up to 10 monomers display a tubular shape, while homologs with 13 or more monomers are locked into a twisted conformation.15–18 Hemicucurbiturils (HCs) are single-bridged CB analogues.19 HC[12], which is formed by the condensation of ethylene urea and formaldehyde and has a highly flexible geometry in solution phase, is the only known 12-membered HC analog.20 The second-largest known HC is the 8-membered, chiral cyclohexanohemicucurbit[8]uril, cycHC[8],21–24 which adopts a tubular shape. Despite detection by mass-spectrometry of cycHCs possessing up to 15 members23,24 and norbornahemicucurbiturils having up to 8 members,25 only smaller homologs have been isolated, namely cycHC[6]s,21,26–29 bambus[n]urils, where n = 4 or 6,30–35 and biotin[6]urils.35–38 These smaller 6-membered homologs adopt a relatively rigid conformation with well-defined cavity shapes.
Herein, we describe the synthesis and isolation of the first enantiopure cyclohexanohemicucurbit[12]uril (cycHC[12]) and detail its conformational dynamics via NMR spectroscopy coupled with density functional theory (DFT) calculations (Fig. 1).
 | ||
| Fig. 1 Chemical structure of cycHC[12] and the lowest energy DFT-modelled (BP86/SVP) structure of cycHC[12]. Blue and green dots represent syn- and anti-methylene bridge configurations, respectively. The blue triangle represents the syn bridge directed inside the cavity. | ||
Our findings show that the macrocycle adopts a concave octagon conformation in toluene at low temperatures, while elevated temperatures result in the activation of two dynamic mechanisms: the bridge flip and the monomer flip. Although cycHC[12] was previously identified by UV-HPLC in a crude reaction mixture,23,24 it was not isolated. We have found that subjecting an isolated macrocycle (cycHC[8]) or the monomer (cyclohexa-1,2-diylurea) to acidic conditions, in the presence of heptafluorobutyric acid (see S2, ESI‡ for details) leads to the formation of longer oligomers and larger macrocycles. The developed procedure produced cycHC[12] with a 1% yield, which permitted elucidation of its shape and conformational dynamics by 1H NMR and DFT. Unfortunately, our attempts to obtain single crystals of cycHC[12] from various solvents have been unsuccessful.
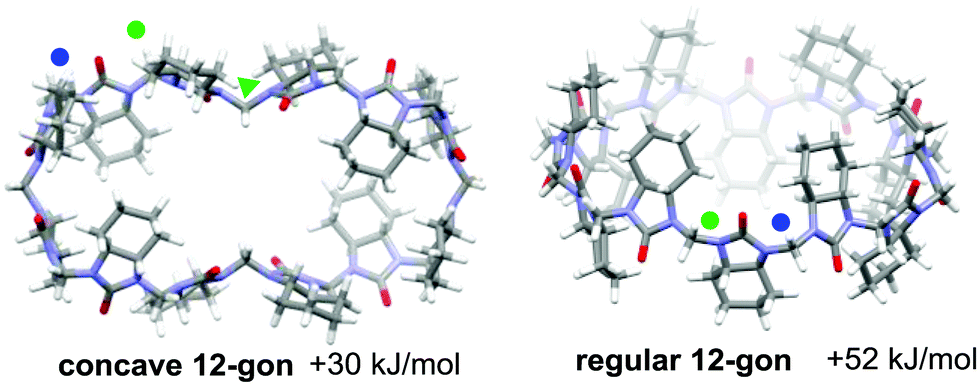
Due to the alternating orientations of the cyclohexano-monomers in HCs, the methylene bridges have two possible configurations: syn or anti. The syn- and anti-configuration describes the mutual orientation of the bridge and its nearest neighboring cyclohexano carbon skeleton – bridges that are pointed toward the same direction as the cyclohexano skeleton are designated as syn. In contrast, those pointed at opposite sides are designated as anti (Fig. 1). These distinct bridges, marked by blue and green colored marks (dots or triangles), are shown in Fig. 1–3. Smaller chiral homologs of cycHCs are known to adopt a barrel-shaped conformation with distinct syn- or anti-oriented bridges in a one-to-one ratio seen in their NMR analysis.21,27 Thus, a similarly regular 12-gon shape was initially expected for the new chiral cycHC[12] macrocycle (Fig. 2, on the right). However, NMR analysis of cycHC[12] in toluene at 265 K did not support a highly symmetric regular 12-gon conformation (Fig. 3). The experimental 1D and 2D NMR spectra of cycHC[12] instead showed the presence of three non-equal urea groups (α, β, and γ) and four methylene bridges (B1, B2, B3, and B4), indicating the existence of two perpendicular axes of C2 symmetry that divide the structure into four symmetry-related parts (Fig. 3A and B). These structural features, supported by NMR data, suggest a concave polygon conformation, with two methylene bridges flipped towards the center of the cavity (Fig. 1, designated with a triangle).
 | ||
| Fig. 2 DFT-modeled structures of cycHC[12] (BP86/SVP) higher energy conformers. Blue and green dots represent syn- and anti-methylene bridge configurations, respectively. The green triangle indicates the anti-bridge directed inside the cavity. | ||
 | ||
| Fig. 3 (A) Symmetry of cycHC[12] and the position of symmetry non-related monomers (α, β, γ). (B) NMR spectra of cycHC[12] bridge protons HB1–HB4 (upper) and carbonyl carbons (lower) in d-toluene. (C) Stacked 13C-NMR spectra of bridge carbons CB1–CB4: (1) experimental at 265 K, (2) experimental at 308 K, (3) simulated for concave octagon, and (4) simulated for regular 12-gon (see simulation details in S20, ESI‡). Blue and green dots designate syn (B1 and B2) and anti (B4) bridge configurations. The blue triangles represent syn-configuration bridges, which are directed inside the cavity (B3). | ||
DFT (BP86/SVP) modeling was used to rationalize the NMR results and investigate the potential cycHC[12] conformers. Three initial geometries were selected, the first based on the barrel-like geometry of smaller homologs and the second and third, concave polygons based on the symmetry revealed by the NMR results. The difference between the two concave structures is the configuration of their inwards-pointing methylene groups. The concave octagon conformer has a syn-oriented inner bridge marked by the blue triangle (Fig. 1), and the concave 12-gon has an anti-oriented inner bridge, marked by the green triangle in (Fig. 2). Of the three optimized structures, the concave octagon conformer is the most prevalent, as it is 52 and 30 kJ mol−1 lower in energy than the regular and concave 12-gon conformers (see S18, ESI‡) respectively. Furthermore, the chemical shifts predicted for the concave octagon conformer (Fig. 3C, spectrum 3) are in good agreement with the experimental values (Fig. 3C, spectrum 1), strongly supporting the presence of this cycHC[12] conformation at 265 K. The calculated 13C chemical shifts of the regular 12-gon conformation (45.7 and 59.4 ppm) (Fig. 3C, spectrum 4) deviate significantly from the experimental cycHC[12] spectra. They more closely resemble those of the barrel-shaped cycHC[8] methylene bridges in similar chiral environments (46.88 ppm and 56.04 ppm) (see S20, ESI‡). In the cycHC[12] experimental 13C NMR spectra, the anti-configuration bridge carbons CB4 resonate at a higher frequency (54.48 ppm) than the CB3 bridge carbons, which are turned inside the cavity and therefore adopt a syn configuration. The latter bridges resonate in a similar range (46.91–47.10 ppm) as the syn-oriented bridges CB1 and CB2 (Fig. 1 and 2). This results from the orientation of CB3 into the cavity, causing its electronic environment, and thus its NMR shielding, to resemble the CB1 and CB2 bridges. In addition, calculations with a higher level of theory, LC-TPSS/aug-pVTZ, were performed to increase the accuracy of the calculated chemical shifts. The addition of long-range corrections made the structures more compact, but the resulting chemical shift values had a greater deviation from the experimental results compared to BP86/SVP (see Table S7, ESI‡). The deviation is due to the compression of cycHC[12], but this is probably not a good representation of the experimental geometry due to the lack of explicit solvent molecules.39
Interestingly, the rotation of the two methylene bridges into the cavity induces a conformational change that strongly affects the localization of the frontier orbitals, concentrating the lowest unoccupied molecular orbital (LUMO) on the urea monomers rather than inside the cavity as observed for cycHC[8] and all smaller HCs28,40 (Fig. 4 and S21, ESI‡).
 | ||
| Fig. 4 LUMO of cycHC[8] and cycHC[12] concave octagons. | ||
Variable temperature (VT) 1H-NMR spectra show the merging of the HB1 signal with the HB2 signal and the HB3 signal with the HB4 signal in pairs between 265 K and 308 K (Fig. 5B, stacked spectra 1 and 6). We suggest this arises from the onset of a dynamic exchange, whereby bridges B1 and B2 and bridges B3 and B4 exchange their magnetic environments in response to the conformational changes of the macrocycle. The merging of the HB1 signal with the HB2 signal and the HB3 signal with the HB4 signal both arise from the same conformational changes in the macrocycle. The bridge flip motion can be best explained as a flip of the B3 bridge outwards and the B4 bridge inwards, which simultaneously exchanges the magnetic environments of the B1 and B2 bridge moieties (Fig. 5A). This process reduces the distance between opposing CB4 methylene bridges from 18 Å to 7 Å while increasing the distance between CB3 bridges. At 265 K, the dynamic exchange is slow, and all bridge environments give separate signals in the 1H NMR spectrum; however, above the coalescence temperature (308 K), the conformations are in fast exchange in the NMR timescale on an 800 MHz spectrometer, resulting in the signals merging (Fig. 5B stacked spectra 6–10).
 | ||
| Fig. 5 (A) Illustration of the cycHC[12] bridge flip mechanism. Dotted arrows designate bridge moieties that move in relation to the center of the macrocycle. Solid arrows designate conformers in equilibrium. Curved arrows designate the conformational flip. Blue and green dots designate syn- and anti-bridge configurations, respectively. The triangle represents the bridges directed into the cavity. (B) Evolution of 1H NMR bridge signals at temperatures from 265 K to 348 K showing the (a) methylene bridge flip and the (b) urea monomer flip. (C) Illustration of monomer flip at higher temperatures at which all bridge signals undergo NMR chemical exchange. On structures cyclohexano groups are omitted for clarity. | ||
At temperatures above 308 K, the two resulting proton signals coalesce, indicating an additional mode of dynamic exchange. We hypothesize that this results from the free rotation of the monomers (Fig. 5B stacked spectra 6–10 and Fig. 5C), averaging the magnetic environments of the syn- and anti-configurations and the inwards and outwards orientations of the bridges. We refer to the second dynamic exchange as the monomer flip. For this process, slow and fast exchange processes are represented by the 308 K and 348 K spectra, respectively (Fig. 5B), and the individual rate constants are available in Table S3 (ESI‡). We undertook line shape analysis of the VT 1H NMR spectra (see Tables S2 and S3 in ESI‡ for fitting details) to derive the activation enthalpy, entropy, and Gibbs free energy values associated with both of the dynamic exchanges (Table 1 and S12, S13, ESI‡). At lower temperatures, the activation Gibbs free energy for the bridge flip is lower than for the monomer flip, which agrees with the monomer flip process having a higher coalescence temperature. It is evident that the bridge flip is more strongly governed by the activation entropy, which we suggest originates from the macrocycle changing its size considerably due to this process.
| Bridge flip | Monomer flip | ||||
|---|---|---|---|---|---|
| ΔH‡ | TΔS‡ | ΔG‡ | ΔH‡ | TΔS‡ | ΔG‡ |
| 38.5 ± 6.1 | −25.5 ± 6.3 | 64.0 ± 8.8 | 59.9 ± 1.1 | −6.6 ± 1.0 | 66.6 ± 1.5 |
In conclusion, we have synthesized, isolated, and characterized the conformation of the largest substituted HC homolog to date. CycHC[12] contains 12 monomers and resembles a concave octagon at 265 K, as revealed by solution NMR analysis and DFT calculations. VT 1H NMR analysis provides evidence for two dynamic processes at elevated temperatures: a bridge flip and a monomer flip. These dynamic processes have not been observed for the cycHC[8] macrocycle due to its small size. We propose that the dynamic characteristics of cycHC[12] would allow it to bind larger species and adapt its structure for a guest molecule by taking advantage of the change in cavity diameter of nearly 1 nm.
The authors would like to acknowledge Oliver Paberits and Karin Kreekman for experimental assistance. We also thank the Estonian MER (Grant No. PRG399 and PSG400), the ERDF (CoE 3.2.0101.08-0017, CoE 2014-2020.4.01.15-0013, and CoE TK134), and H2020-FETOPEN (828779 (INITIO)) for financial support.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- L. Cronin, P. A. McGregor, S. Parsons, S. Teat, R. O. Gould, V. A. White, N. J. Long and N. Robertson, Inorg. Chem., 2004, 43, 8023–8029 CrossRef CAS.
- X. Chi, G. Yu, L. Shao, J. Chen and F. Huang, J. Am. Chem. Soc., 2016, 138, 3168–3174 CrossRef CAS.
- W. Saenger, J. Jacob, K. Gessler, T. Steiner, D. Hoffmann, H. Sanbe, K. Koizumi, S. M. Smith and T. Takaha, Chem. Rev., 1998, 98, 1787–1802 CrossRef CAS.
- K. Gessler, I. Usón, T. Takaha, N. Krauss, S. M. Smith, S. Okada, G. M. Sheldrick and W. Saenger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4246–4251 CrossRef CAS.
- H. Taira, H. Nagase, T. Endo and H. Ueda, J. Inclusion Phenom. Macrocyclic Chem., 2006, 56, 23–28 CrossRef CAS.
- M. G. Gotsev and P. M. Ivanov, ARKIVOC, 2007, 13, 167–189 Search PubMed.
- K. I. Assaf, D. Gabel, W. Zimmermann and W. M. Nau, Org. Biomol. Chem., 2016, 14, 7702–7706 RSC.
- C. Sonnendecker, S. Thürmann, C. Przybylski, F. D. Zitzmann, N. Heinke, Y. Krauke, K. Monks, A. A. Robitzki, D. Belder and W. Zimmermann, Angew. Chem., Int. Ed., 2019, 58, 6411–6414 CrossRef CAS.
- K. Harata, T. Endo, H. Ueda and T. Nagai, Supramol. Chem., 1998, 9, 143–150 CrossRef CAS.
- J. Jacob, K. Geßler, D. Hoffmann, H. Sanbe, K. Koizumi, S. M. Smith, T. Takaha and W. Saenger, Angew. Chem., Int. Ed., 1998, 37, 605–609 CrossRef.
- X.-B. Hu, Z. Chen, L. Chen, L. Zhang, J.-L. Hou and Z.-T. Li, Chem. Commun., 2012, 48, 10999–11001 RSC.
- V. Guérineau, M. Rollet, S. Viel, B. Lepoittevin, L. Costa, P. Saint-Aguet, R. Laurent, P. Roger, D. Gigmes, C. Martini and V. Huc, Nat. Commun., 2019, 10, 1–14 CrossRef.
- C. Bavoux, R. Baudry, I. Dumazet-Bonnamour, R. Lamartine and M. Perrin, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 221–224 CrossRef CAS.
- Q. Li, S.-C. Qiu, J. Zhang, K. Chen, Y. Huang, X. Xiao, Y. Zhang, F. Li, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, Z. Tao, L. F. Lindoy and G. Wei, Org. Lett., 2016, 18, 4020–4023 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- W.-H. Huang, S. Liu, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14744–14745 CrossRef CAS.
- X.-J. Cheng, L.-L. Liang, K. Chen, N.-N. Ji, X. Xiao, J.-X. Zhang, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, X.-L. Ni and Z. Tao, Angew. Chem., Int. Ed., 2013, 52, 7252–7255 CrossRef CAS.
- N. N. Andersen, M. Lisbjerg, K. Eriksen and M. Pittelkow, Isr. J. Chem., 2018, 58, 435–448 CrossRef CAS.
- Y. Miyahara, K. Goto, M. Oka and T. Inazu, Angew. Chem., Int. Ed., 2004, 43, 5019–5022 CrossRef CAS.
- E. Prigorchenko, M. Oeren, S. Kaabel, M. Fomitsenko, I. Reile, I. Jarving, T. Tamm, F. Topic, K. Rissanen and R. Aav, Chem. Commun., 2015, 51, 10921–10924 RSC.
- S. Kaabel, J. Adamson, F. Topić, A. Kiesilä, E. Kalenius, M. Öeren, M. Reimund, E. Prigorchenko, A. Lõokene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- M. Fomitšenko, E. Shmatova, M. Öeren, I. Järving and R. Aav, Supramol. Chem., 2014, 26, 698–703 CrossRef.
- S. Kaabel, R. S. Stein, M. Fomitšenko, I. Järving, T. Friščić and R. Aav, Angew. Chem., Int. Ed., 2019, 58, 6230–6234 CrossRef CAS.
- T. Fiala and V. Sindelar, Synlett, 2013, 24, 2443–2445 CrossRef CAS.
- Y. Li, L. Li, Y. Zhu, X. Meng and A. Wu, Cryst. Growth Des., 2009, 9, 4255–4257 CrossRef CAS.
- R. Aav, E. Shmatova, I. Reile, M. Borissova, F. Topić and K. Rissanen, Org. Lett., 2013, 15, 3786–3789 CrossRef CAS.
- M. Öeren, E. Shmatova, T. Tamm and R. Aav, Phys. Chem. Chem. Phys., 2014, 16, 19198–19205 RSC.
- E. Prigorchenko, S. Kaabel, T. Narva, A. Baškir, M. Fomitšenko, J. Adamson, I. Järving, K. Rissanen, T. Tamm and R. Aav, Chem. Commun., 2019, 55, 9307–9310 RSC.
- O. Reany, A. Mohite and E. Keinan, Isr. J. Chem., 2018, 58, 449–460 CrossRef CAS.
- T. Lizal and V. Sindelar, Isr. J. Chem., 2018, 58, 326–333 CrossRef CAS.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS.
- M. A. Yawer, V. Havel and V. Sindelar, Angew. Chem., Int. Ed., 2015, 54, 276–279 CAS.
- V. Havel, M. Babiak and V. Sindelar, Chem. – Eur. J., 2017, 23, 8963–8968 CrossRef CAS.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000–4003 CrossRef CAS.
- M. Lisbjerg, B. M. Jessen, B. Rasmussen, B. E. Nielsen, A. Ø. Madsen and M. Pittelkow, Chem. Sci., 2014, 5, 2647–2650 RSC.
- M. Lisbjerg, B. E. Nielsen, B. O. Milhøj, S. P. A. Sauer and M. Pittelkow, Org. Biomol. Chem., 2015, 13, 369–373 RSC.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS.
- M. A. Iron, J. Chem. Theory Comput., 2017, 13, 5798–5819 CrossRef CAS.
- T. F. G. G. Cova, S. C. C. Nunes, A. J. M. Valente, T. M. V. D. Pinho e Melo and A. A. C. C. Pais, J. Mol. Liq., 2017, 242, 640–652 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of late Prof. Hans J. Reich. |
| ‡ Electronic supplementary information (ESI) available: Synthesis, isolation, characterization of cycHC[12] by HRMS, 1D and 2D NMR, VT-NMR, conformational dynamics analysis, DFT calculations is described. See DOI: 10.1039/d0cc06817a |
| This journal is © The Royal Society of Chemistry 2020 |