
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Creation of active water-splitting photocatalysts by controlling cocatalysts using atomically precise metal nanoclusters
Tokuhisa
Kawawaki
 abc,
Yuki
Kataoka
a,
Shuhei
Ozaki
a,
Masanobu
Kawachi
a,
Momoko
Hirata
a and
Yuichi
Negishi
*abc
abc,
Yuki
Kataoka
a,
Shuhei
Ozaki
a,
Masanobu
Kawachi
a,
Momoko
Hirata
a and
Yuichi
Negishi
*abc
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
bResearch Institute for Science and Technology, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
cPhotocatalysis International Research Center, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
First published on 8th December 2020
Abstract
With global warming and the depletion of fossil resources, our fossil-fuel-dependent society is expected to shift to one that instead uses hydrogen (H2) as clean and renewable energy. Water-splitting photocatalysts can produce H2 from water using sunlight, which are almost infinite on the earth. However, further improvements are indispensable to enable their practical application. To improve the efficiency of the photocatalytic water-splitting reaction, in addition to improving the semiconductor photocatalyst, it is extremely effective to improve the cocatalysts (loaded metal nanoclusters, NCs) that enable the reaction to proceed on the photocatalysts. We have thus attempted to strictly control metal NCs on photocatalysts by introducing the precise-control techniques of metal NCs established in the metal NC field into research on water-splitting photocatalysts. Specifically, the cocatalysts on the photocatalysts were controlled by adsorbing atomically precise metal NCs on the photocatalysts and then removing the protective ligands by calcination. This work has led to several findings on the electronic/geometrical structures of the loaded metal NCs, the correlation between the types of loaded metal NCs and the water-splitting activity, and the methods for producing high water-splitting activity. We expect that the obtained knowledge will lead to clear design guidelines for the creation of practical water-splitting photocatalysts and thereby contribute to the construction of a hydrogen-energy society.
 Tokuhisa Kawawaki | Tokuhisa Kawawaki Assistant Professor of the Department of Applied Chemistry at Tokyo University of Science. He received a PhD degree (2015) in Applied Chemistry from the University of Tokyo. In 2016–2017, he worked as a Japan Society for the Promotion of Science (JSPS) Postdoctoral fellow (PD) at the University of Melbourne. In 2017–2018, he worked as a JSPS super PD (SPD) at Kyoto University. In 2019, he moved to his current position. His current research topics include the synthesis of metal nanoparticles and nanoclusters in solution and their applications for photoelectrochemistry and photocatalysts. |
 Yuki Kataoka | Yuki Kataoka Master course student in the Negishi group at Tokyo University of Science. He received a BSc (2019) in Chemistry from Tokyo University of Science. His research interests include the activation of water-splitting photocatalysts by heteroatom doping of cocatalysts and elucidation of the calcination process for loading precise metal NCs on photocatalysts. |
 Shuhei Ozaki | Shuhei Ozaki Master course student in the Negishi group at Tokyo University of Science. He received a BSc (2019) in Chemistry from Tokyo University of Science. In 2019, he worked as a study abroad student at The University of Adelade (Greg Metha's group). His research interests include the activation of visible-light-driven water-splitting photocatalysts. |
 Masanobu Kawachi | Masanobu Kawachi Master course student in the Negishi group at Tokyo University of Science. He received a BSc (2020) in Chemistry from Tokyo University of Science. His research interests include the activation of visible-light-driven water-splitting photocatalysts. |
 Momoko Hirata | Momoko Hirata Master course student in the Negishi group at Tokyo University of Science. She received a BSc (2020) in Chemistry from Tokyo University of Science. Her research interests include the elucidation of the calcination process for loading precise metal NCs on photocatalysts. |
 Yuichi Negishi | Yuichi Negishi Professor of the Department of Applied Chemistry at Tokyo University of Science. He received a PhD degree in Chemistry in 2001 under the supervision of Prof. Atsushi Nakajima from Keio University. Before joining Tokyo University of Science in 2008, he was employed as an Assistant Professor at Keio University and at the Institute for Molecular Science. His current research interests include the precise synthesis of stable and functionalized metal nanoclusters and their applications in energy and environmental materials. |
1. Introduction
1.1. Background
In the 21st century, humankind is facing unprecedented serious energy and environmental issues such as the depletion of fossil resources and the destruction of the environment on a global scale.1 Thus, it is expected to address these problems to create a sustainable society as soon as possible. The energy conversion system shown in Fig. 1 is one of the ultimate systems for constructing such a society.2 In this system, hydrogen (H2) is produced by a photocatalyst3 and/or an electrolysis cell,4–15 and the obtained H2 is converted into electric power by a fuel cell.16–24 With such a system, it is possible to obtain energy from water and sunlight, which are almost infinite on the earth. In addition, when this system is used, the energy medium (H2) can be circulated, preventing the problem of energy depletion. Furthermore, carbon dioxide (CO2), which leads to global warming, is not generated in this system. For constructing this system, modern chemistry is expected to improve the functionality of the photocatalysts, electrolysis cells, and fuel cells.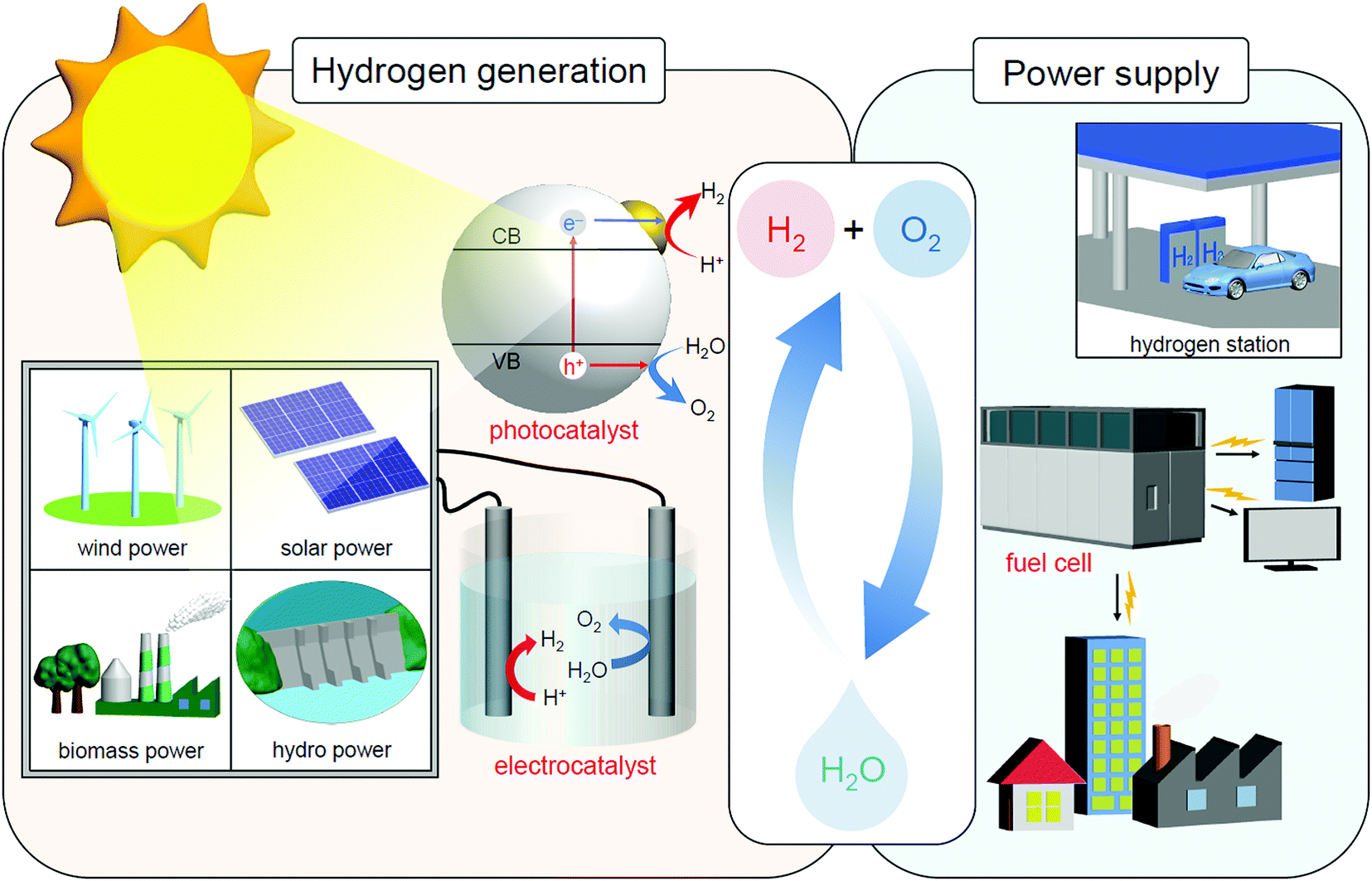 | ||
| Fig. 1 Schematic of the energy conversion system expected for constructing a sustainable society. Note that sunlight also produces wind, biomass, and hydro power in addition to solar power. | ||
1.2. Water-splitting photocatalysts
For H2 production, photocatalytic reactions (Fig. 1) can produce H2 directly from water and sunlight.25–37 Therefore, the photocatalytic water-splitting reaction is considered to be one of the cleanest energy-production reactions for humankind. Among the materials that enable such a reaction to proceed, powder semiconductor photocatalysts have the advantages of a simple system and the ease of increasing the surface area. Because of these advantages, if a large-scale H2-production plant using a powder semiconductor photocatalyst is constructed in a desert or uninhabited island where sunlight is abundant, a large amount of H2 is expected to be generated.38 However, currently, the solar-to-hydrogen (STH) conversion efficiency is only 1.1%.39 To enable practical use of a water-splitting photocatalyst, it is indispensable to improve the STH conversion efficiency to approximately 5–10%.33When a semiconductor photocatalyst is used, H2 and oxygen (O2) are produced from water via the following three processes:25 (i) excitation of electrons from the valence band (VB) to the conduction band (CB) of the photocatalyst by light irradiation, (ii) charge transfer of excited electrons and holes to the surface of the photocatalyst, and (iii) reduction and oxidation of water by excited electrons and holes, respectively (Fig. 2A). Theoretically, when the bottom edge of the CB of the semiconductor photocatalyst is on the negative side of the reduction potential of water (0 V vs. normal hydrogen electrode (NHE)), the reduction reaction of water proceeds (Fig. 2B) and H2 is produced (hydrogen evolution reaction, HER; Fig. 2A). If the top edge of the VB of the semiconductor photocatalyst is on the positive side of the oxidation potential of water (1.23 V vs. NHE), the oxidation reaction of water proceeds (Fig. 2B) and O2 is generated (oxygen evolution reaction, OER; Fig. 2A). However, even a semiconductor photocatalyst that satisfies these conditions cannot necessarily achieve complete water splitting because of the high activation energy in each reaction. Therefore, to decrease the activation energy of the reaction, metal and/or metal oxide nanoparticles (NPs) made of precious metals are generally loaded on the photocatalyst as active sites (cocatalyst; Fig. 2A).2,27 This loading of cocatalysts also promotes the separation of electrons and holes in the photocatalyst, thereby suppressing the deactivation of the reaction based on the recombination of electrons and holes. Thus, to enhance the efficiency of the water-splitting reaction, it is extremely effective to improve the cocatalyst in addition to improving the semiconductor photocatalysts.38
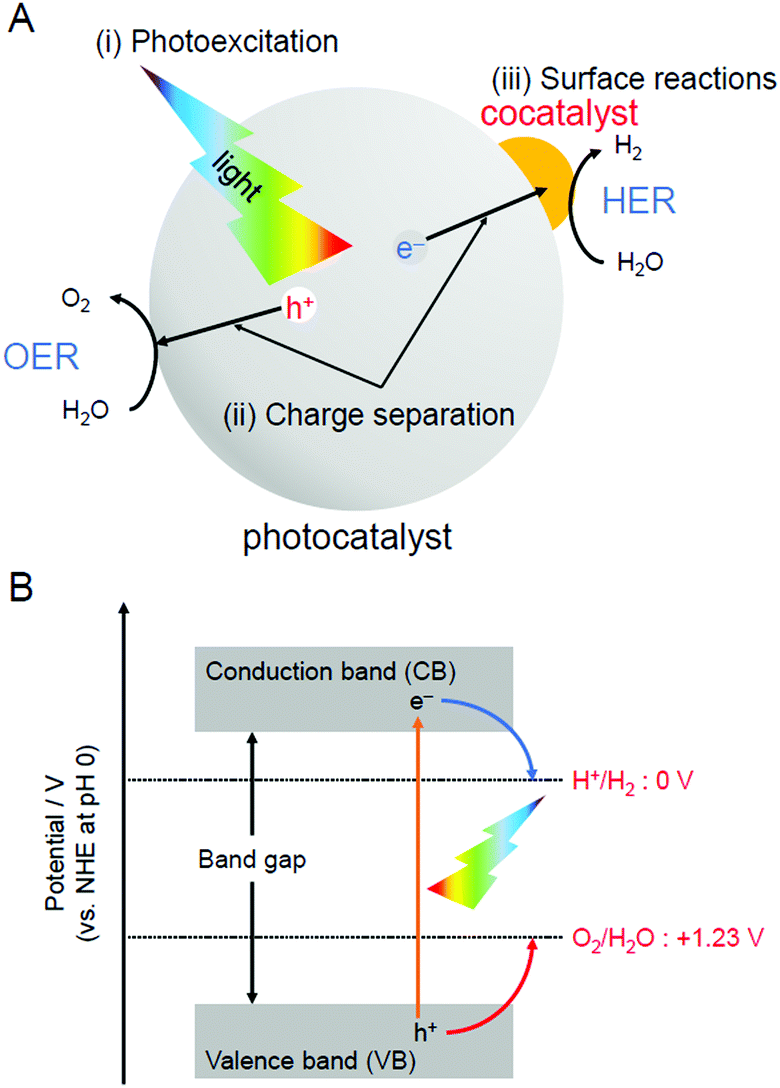 | ||
| Fig. 2 (A) Schematic of photocatalytic reactions: (i) photoexcitation, (ii) charge separation, and (iii) surface reactions. (B) Relationship between the semiconductor band structure and redox potentials of water splitting. | ||
1.3. Control of cocatalysts
Chemical reactions occurring on the surface of the cocatalyst involve the adsorption and reaction of reactants and desorption of products. Therefore, the efficiency of the chemical reaction is greatly affected by the adsorption/desorption energy of the reactants/products on the surface of the cocatalyst. Previous studies have revealed that the chemical reaction on the surface of the cocatalyst tends to proceed easily when the Gibbs energy in the process of adsorption/desorption of the reactants/products is moderate. For this reason, metal/metal oxide NPs of group 8–11 elements with sizes of several to several tens of nanometers are generally used as cocatalysts.2In many cases, the cocatalysts are loaded using the impregnation (Fig. 3A)35 or photodeposition method (Fig. 3B).40 These methods enable the cocatalyst to be loaded on the photocatalyst using a very simple procedure. When the photodeposition method is used, it is also possible to preferentially load the cocatalyst on the specific crystal planes of the photocatalysts suitable for the reaction.41 Through studies using these loading methods, it has been clarified that miniaturization of cocatalysts42 and control of the electronic structure by alloying43 are extremely effective in creating highly functional water-splitting photocatalysts.
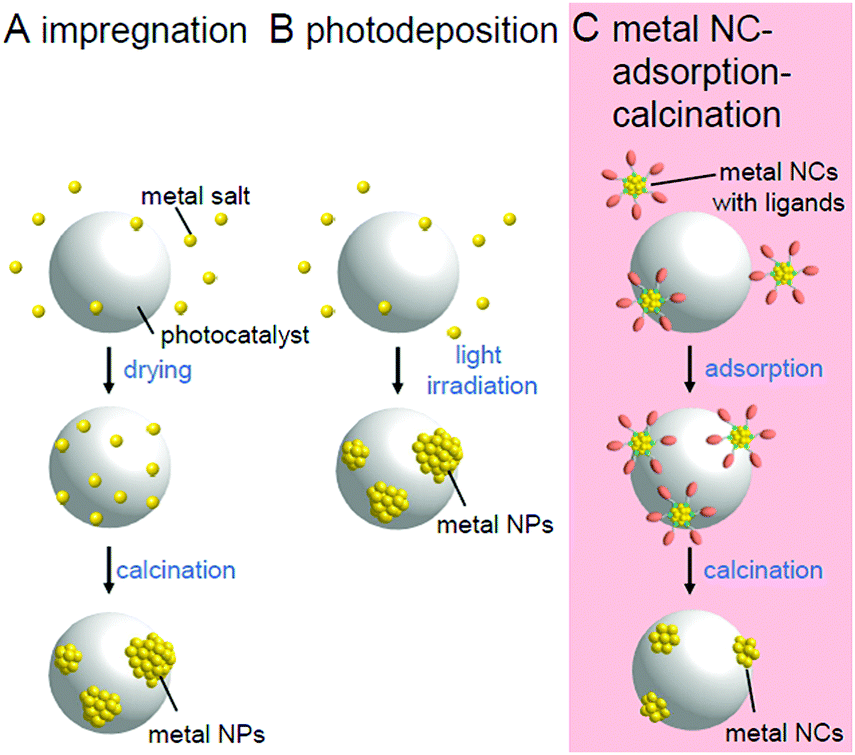 | ||
| Fig. 3 Comparison of the (A) impregnation, (B) photodeposition, and (C) metal NC adsorption–calcination methods. | ||
However, the electronic structure of fine metal/metal oxide nanoclusters (NCs) varies greatly depending on the number of constituent atoms and chemical compositions.44–65 Therefore, if the chemical composition of the fine cocatalysts can be controlled with atomic precision, it is possible to create highly active photocatalysts with the selective loading of a highly active cocatalyst on the surface. In addition, for metal NCs loaded with atomic precision, it is possible to obtain a deep understanding of the electronic/geometrical structures of the cocatalysts and the interaction between the photocatalyst surface and cocatalysts through various high-resolution measurements and theoretical calculations.66–77 Therefore, the strict control of the cocatalyst is expected to provide deep understanding of the key factors toward high activation. If we would design and create appropriate metal NCs based on the attained information, great enhancement of the water-splitting activities of the photocatalysts could be achieved.78 Metal NCs can be synthesized with atomic precision using thiolates (SR),21,79–114 selenolates (SeR),115–122 phosphines (PR3), alkynes (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR),123,124 carbon monoxide (CO), dendrimers,125etc. as protective ligands.126–143 Furthermore, previous studies have demonstrated that size-controlled metal NCs can be loaded on a support by adsorbing such precise metal NCs on the support and then removing the protective ligands by calcination (Fig. 3C).144–146
CR),123,124 carbon monoxide (CO), dendrimers,125etc. as protective ligands.126–143 Furthermore, previous studies have demonstrated that size-controlled metal NCs can be loaded on a support by adsorbing such precise metal NCs on the support and then removing the protective ligands by calcination (Fig. 3C).144–146
1.4. Contents of this review
We aim to improve the functionality of water-splitting photocatalysts to a practical level (Fig. 4) and thereby contribute to the construction of a hydrogen-energy society.2 To achieve this, we have attempted to strictly control metal NCs on photocatalysts and thereby clarify the details of the effect of controlling the cocatalyst on the water-splitting activity by introducing the precise-control techniques of metal NCs established in the metal NC field into research on water-splitting photocatalysts.147,148 This feature article summarizes our previous findings.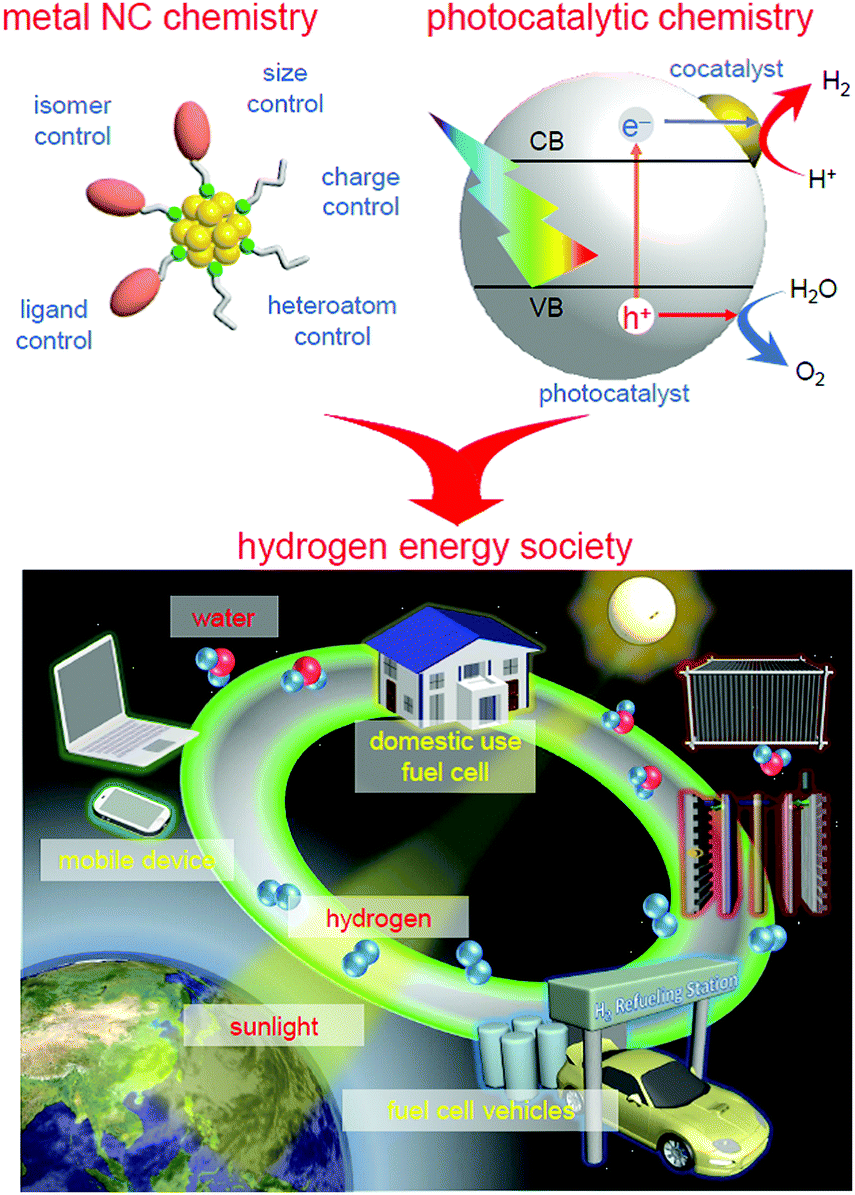 | ||
| Fig. 4 Schematic of the aims of our study. | ||
In Section 2, we first describe the synthesis method and geometrical structure of metal NCs controlled with atomic precision that are used as precursors of cocatalysts. Then, in Section 3, we describe our research on the functionalization of water-splitting photocatalysts using the metal NCs as precursors of cocatalysts. Specifically, the precise loading of the metal NCs on a photocatalyst (Section 3.1), the electronic/geometrical structure of the loaded metal NCs (Section 3.2), the correlation between the types of loaded metal NCs and the water-splitting activity (Section 3.3), and the functionalization of photocatalysts (Section 3.4) are described. After a brief summary in Section 4, the future outlook is described in Section 5.
2. Ligand-protected atomically precise metal NCs
Since the 1970s, there have been many reports on the precise synthesis of metal NCs using PR3, CO, and halogens as ligands.126–143 [Au11(PPh3)7I3]+ (Au = gold; PPh3 = triphenylphosphine; I = iodine),149 Au55(PPh3)12Cl6 (Schmid's Au55 cluster; Cl = chlorine),150 and [Pt3n(CO)6n]2− (Chini cluster; Pt = platinum)151,152 are representative examples of these metal NCs. However, since 2000, research on metal NCs protected by SR, SeR, and/or CCR, which are relatively easily synthesized and can be handled in the atmosphere, has been actively conducted. Herein, we only provide an outline of these metal NCs because several groups including us have already published many reviews on these metal NCs.72,93,94,153–161
2.1. Synthesis methods
Fig. 5 presents a typical method for precisely synthesizing SR-protected metal NCs.93,148 In the method shown in Fig. 5A, metal NCs are prepared by reducing a metal salt or a metal–SR complex with a reducing agent.162 The metal NCs obtained by this method normally have a distribution in their chemical compositions. Therefore, each metal NC with atomic precision can be obtained by the separation of the mixture using fractionation techniques such as polyacrylamide gel electrophoresis (PAGE),79,163–168 high-performance liquid chromatography (HPLC),55,156,169–182 and thin-layer chromatography (TLC).183–185 As opposed to separating them, when the mixture is exposed to severe conditions, less stable species transform into stable species, thereby allowing stable metal NCs to be size-selectively synthesized (size focusing; Fig. 5B).153,186–188 In the method shown in Fig. 5C, metal NCs are grown with relatively uniform size by selecting appropriate experimental conditions for slowly reducing the metal ions and aggregating the obtained metal atoms (slow reduction).189–192 In the method shown in Fig. 5D, metal NCs with a certain chemical composition are first selectively synthesized, and then the ligands are replaced with ligands with a significantly different structure. This reaction finally results in the selective formation of metal NCs with different chemical compositions (transformation).193 When using another type of metal salts, metal–SR complexes, or metal NCs as a reactant, it is also possible to selectively synthesize alloy NCs composed of several elements (reconstruction).194,195 In the method shown in Fig. 5E, after selectively synthesizing metal NCs with a certain chemical composition, the metal NCs are reacted with metal salts,196 metal–SR complexes,197 metal NCs,198 or a metal plate199 including other types of metal to replace some metal atoms with other metal elements (metal exchange). Under particular reaction conditions, such metal exchange does not occur, and another type of metal atoms or metal ions are deposited on the surface of the metal NCs (deposition of metal atoms/ions; Fig. 5F).184,200,201 In these synthetic methods, the method shown in Fig. 5A is superior for systematically isolating a series of metal NCs, and the method shown in Fig. 5B–F is superior for size-selectively synthesizing a specific metal NC. In addition to these methods, there have been several reports on the synthesis of new metal NCs using a combination of several of the methods shown in Fig. 5.202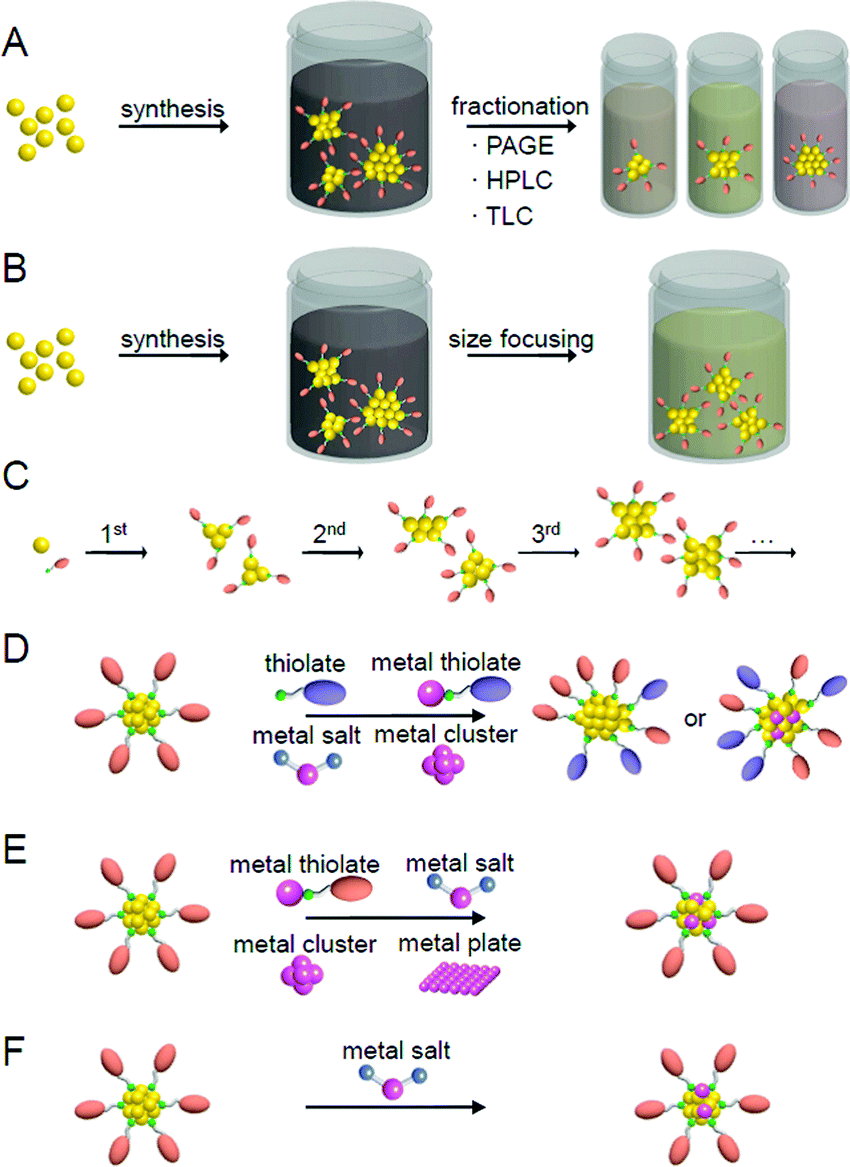 | ||
| Fig. 5 Typical methods for the precise synthesis of metal clusters: (A) fractionation, (B) size focusing, (C) slow reduction, (D) transformation/reconstruction, (E) metal exchange, and (F) deposition of metal atoms/ions. | ||
2.2. Geometrical structures
When research on SR-protected metal NCs began, they were thought to have a geometrical structure where the metal core is covered by SR.203 However, the density functional theory (DFT) calculations on [Au38(SCH3)24]0 by Häkkinen et al. in 2006204 suggested that SR-protected metal NCs have a geometrical structure where the metal core is covered by Au–SR oligomers (now called staples). In the next year, this “divide and protect” structure was experimentally verified by Kornberg et al. through single-crystal X-ray diffraction (SC-XRD) analysis of [Au102(p-MBA)44]0 (p-MBA = p-mercaptobenzoic acid) (Fig. 6A).111,205 Since then, the geometrical structures of many SR-protected metal NCs have been determined by SC-XRD,153,154 and almost all of the SR-protected metal NCs have been observed to have such “divide and protect” structures (Fig. 6B–D). Several research groups, including those of Zhu, Wang, Jiang, Tsukuda, and Pei et al., have shown that SeR-protected metal NCs (Fig. 6E)206,207 and CCR-protected metal NCs (Fig. 6F)124,208,209 also have such “divide and protect” framework structures.
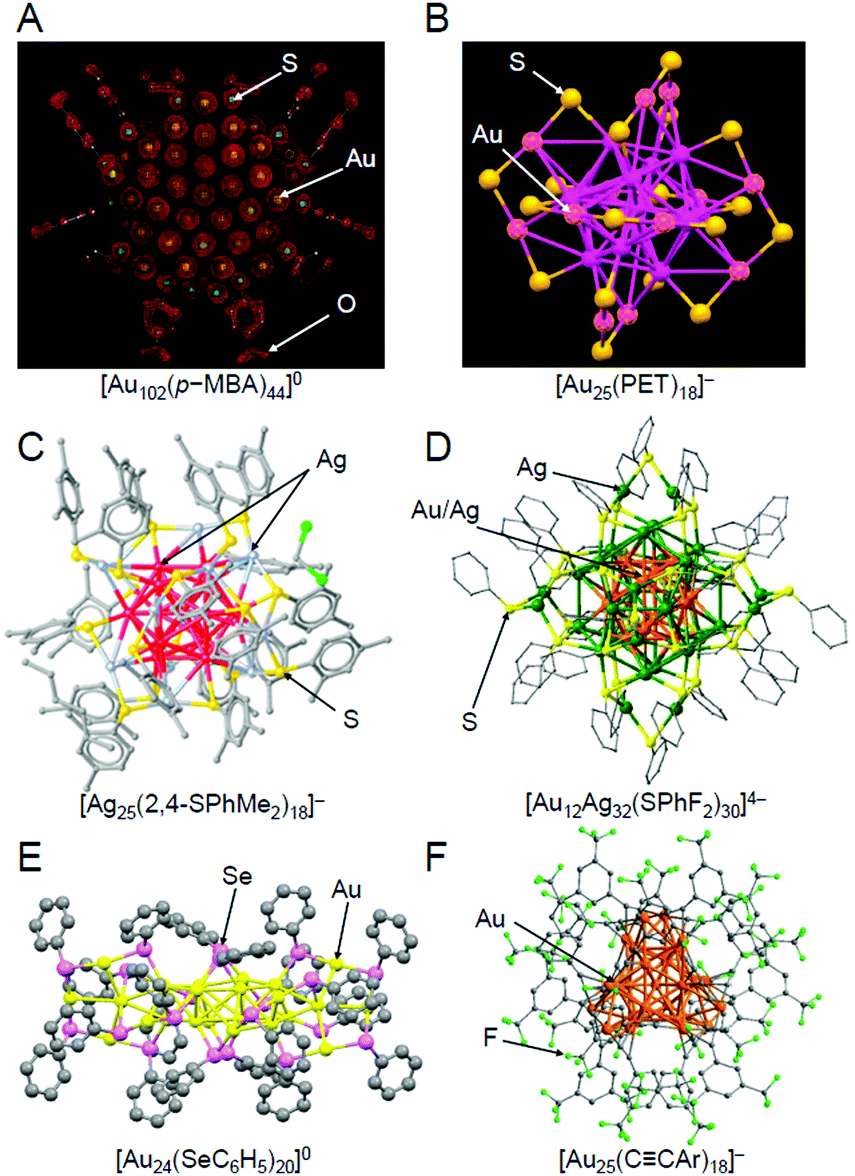 | ||
| Fig. 6 Geometric structures of (A) [Au102(p-MBA)44]0,111 (B) [Au25(PET)18]− (PET = phenylethanethiolate),112 (C) [Ag25(2,4-SPhMe2)18]− (2,4-SPhMe2 = 2,4-dimethylbenzenethiolate),113 (D) [Au12Ag32(SPhF2)30]4− (SPhF2 = 3,4-difluorothiophenol),114 (E) [Au24(SeC6H5)20]0 (SeC6H5 = benzeneselenolate),206 and (F) [Au25(CCAr)18]− (CCAr = 3,5-bis(trifluoromethyl)phenylethynyl).124 The gray points represent carbon atoms. Hydrogen atoms are not shown. Au25(SG)18, which was used in our study, has been considered to have a similar framework structure to [Au25(PET)18]− (B), although its geometric structure has not been determined by SC-XRD to date. Reproduced with permission from ref. 111, 112, 113, 114, 206 and 124. Copyright 2007 American Association for the Advancement of Science, Copyright 2008 American Chemical Society, Copyright 2015 American Chemical Society, Copyright 2013 Springer Nature Communications, Copyright 2014 American Chemical Society, and Copyright 2019 Wiley-VCH, respectively. | ||
3. Activation of water-splitting photocatalysts using atomically precise metal NCs
We aim to functionalize water-splitting photocatalysts that can be used as practical materials. Therefore, we used some of the most advanced photocatalysts. Fig. 7 shows the band structures of several advanced photocatalysts.28,29,210,211 Among them, BaLa4Ti4O15 (Fig. 8A)210 or SrTiO3 (Fig. 8B)212,213 were used in our research. BaLa4Ti4O15 is a photocatalyst210 developed by Kudo et al. in 2009 and has a perovskite structure (Fig. 8A) (band gap = 3.8 eV). Because this photocatalyst is not commercially available, BaLa4Ti4O15 was synthesized in our laboratory using a previously reported procedure. SrTiO3 also has a perovskite structure with a band gap of 3.2 eV (Fig. 8B). The band gap of this photocatalyst can be tuned by doping with different elements (rhodium (Rh), lanthanum (La), etc.) and the obtained SrTiO3 doped with different elements (SrTiO3:Rh, SrTiO3:La, Rh, etc.) has been used as a photocatalyst for H2 evolution in Z-scheme systems.26,31,214–217 Because SrTiO3 is already on the market, we used a commercial product for this photocatalyst.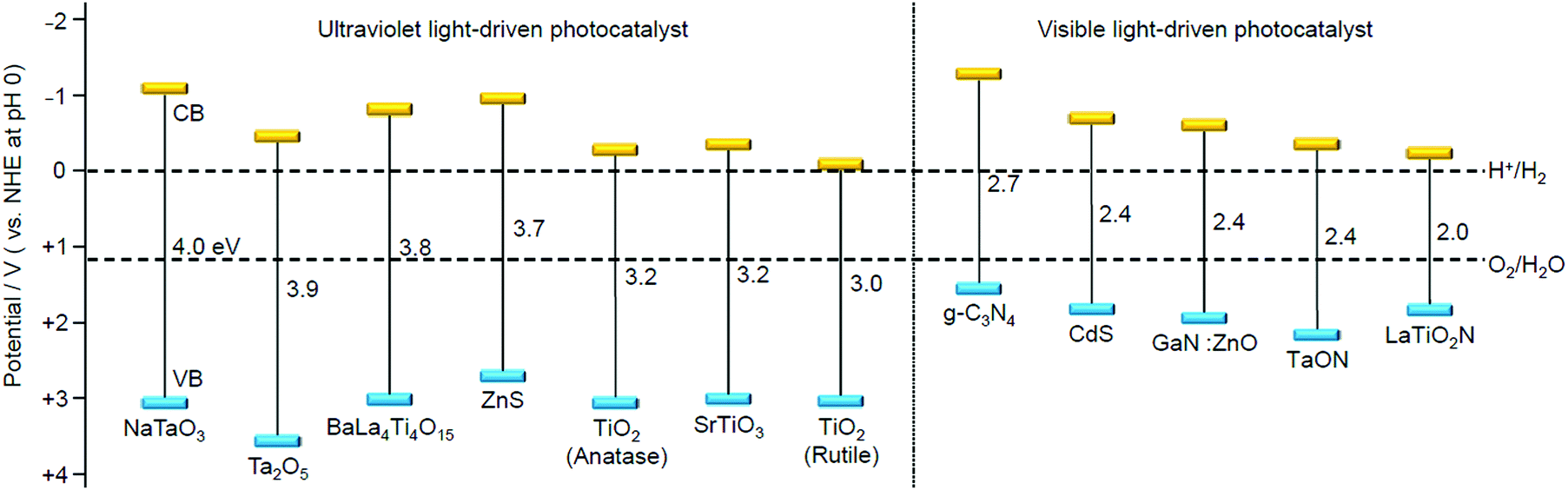 | ||
| Fig. 7 Energy-band diagram of several water-splitting photocatalysts and the redox potential of the hydrogen- and oxygen-evolution reactions.28,29,210,211 | ||
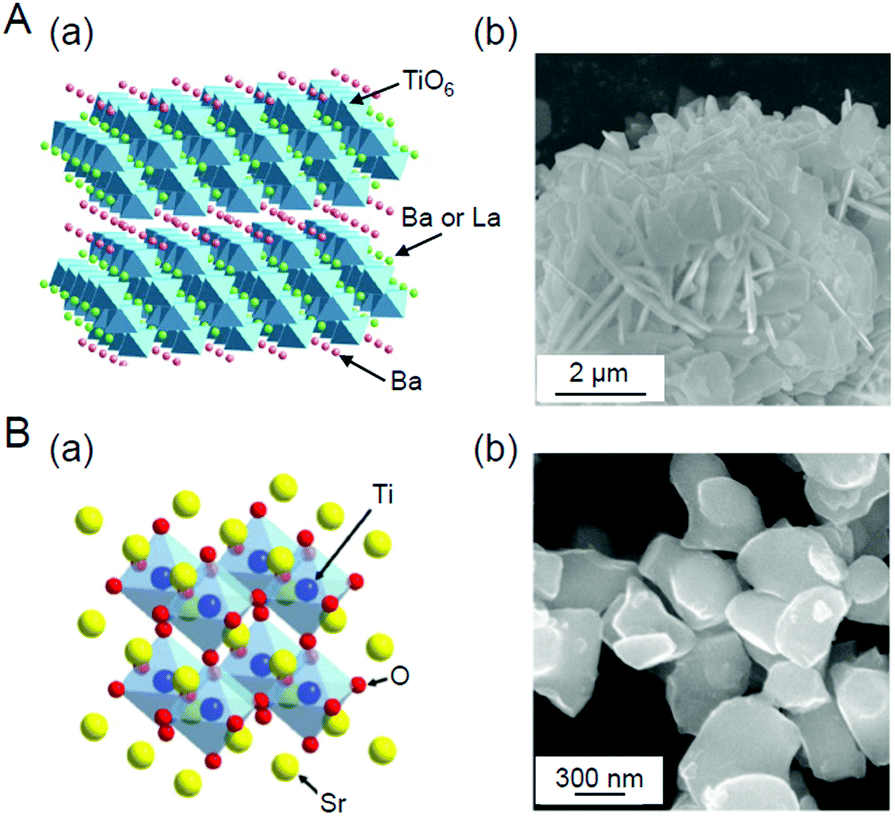 | ||
| Fig. 8 (A) (a) Geometric structure of BaLa4Ti4O15247 and (b) scanning electron microscope (SEM) image of the prepared BaLa4Ti4O15.270 (B) (a) Geometric structure of SrTiO3 and (b) SEM image of the prepared SrTiO3.231 Reproduced with permission from ref. 247 and 270. Copyright 2018 American Chemical Society, Copyright 2020 Wiley-VCH. | ||
3.1. Loading of atomically precise metal NCs on photocatalysts
When a metal oxide is added to water, hydroxyl groups (–OH) are normally formed on the surface. Hydrophilic metal NCs with proton functional groups are easily adsorbed on such surfaces.146,218–222 Therefore, we first worked on loading metal NCs on BaLa4Ti4O15 using hydrophilic metal NCs as precursors.Loading of Au25. In our first effort, we used glutathionate (SG)-protected Au25 NCs (Au25(SG)18; Fig. 6B) as a precursor, which are stable in solution and can be size-selectively synthesized.79,187,223,224 In this experiment, Au25(SG)18 was first adsorbed on BaLa4Ti4O15 by stirring them in water (Au25(SG)18/BaLa4Ti4O15).225 In the optical absorption spectrum of the aqueous solution after adsorption, optical absorption was hardly observed, indicating that almost all of the Au25(SG)18 was adsorbed on BaLa4Ti4O15. Glutathionate has two carboxyl groups and one amino group.226 It can be considered that Au25(SG)18 was efficiently adsorbed on the photocatalyst because the polar functional groups of Au25(SG)18 formed hydrogen bonds with the –OH groups on the surface of BaLa4Ti4O15.
Then, the obtained Au25(SG)18/BaLa4Ti4O15 was calcined in an electric furnace. Thermogravimetric analysis (TGA) of Au25(SG)18/BaLa4Ti4O15 showed that most of the SG can be removed at 300 °C.225 Thus, Au25(SG)18/BaLa4Ti4O15 was calcined at 300 °C for 2 h under reduced pressure. Fig. 9A–D present the transmission electron microscope (TEM) images, S 2p and Au 4f spectra obtained by X-ray photoelectron spectroscopy (XPS), and optical adsorption/diffuse reflectance spectrum of the photocatalysts before and after calcination (Au25(SG)18/BaLa4Ti4O15 and Au25/BaLa4Ti4O15), respectively, for the photocatalysts containing Au with a weight ratio of 0.1 wt% (0.1 wt% Au). In the TEM image of the sample after calcination (Fig. 9A(c)), only particles with sizes similar to those before calcination were observed, indicating that calcination did not cause the aggregation of NCs. In the S 2p spectrum (Fig. 9B(b)), a peak was hardly observed, indicating that most of the ligands were removed by calcination. In the Au 4f spectrum of the sample after calcination, the peak positions (83.7 and 87.7 eV; Fig. 9C(b)) were different from those of the sample before calcination (84.7 and 88.5 eV; Fig. 9C(a)) and were located at an energy near those expected for Au(0) (84.0 and 87.7 eV). For Au25(SG)18, the Au 4f peaks appear on the oxidation side (high-energy side) compared with Au(0) because of the partial charge transfer from Au to S.225 The shift of the Au 4f peaks after calcination strongly indicates that the ligands were removed by calcination and therefore that Au in the metallic state was loaded on BaLa4Ti4O15. In the diffuse reflectance spectrum of the photocatalyst after calcination (Fig. 9D(c)), the peaks in the visible region, which are characteristic of Au25(SG)18, were not observed. In the optical absorption spectrum of Au25(SR)18 (Fig. 9D(a)), the peaks in the region of 600–800 nm are attributed to the absorption originating in the central Au13 core of Au25(SR)18 (Fig. 6B).112,227–230 It can be interpreted that these peaks disappeared because the ligands were removed by calcination, thereby resulting in a drastic change of the structure of the NCs. The slight increase in the particle size of the photocatalysts after calcination (Fig. 9A) can also be explained by this structural change. These results confirmed that the ligands were removed from Au25(SG)18/BaLa4Ti4O15 by calcination while maintaining the particle size of the Au25 NC (Au25/BaLa4Ti4O15).225 In our later research, it became clear that the precise loading of Au25 using Au25(SG)18 as a precursor can also be performed in the same way when SrTiO3 is used as a photocatalyst.231
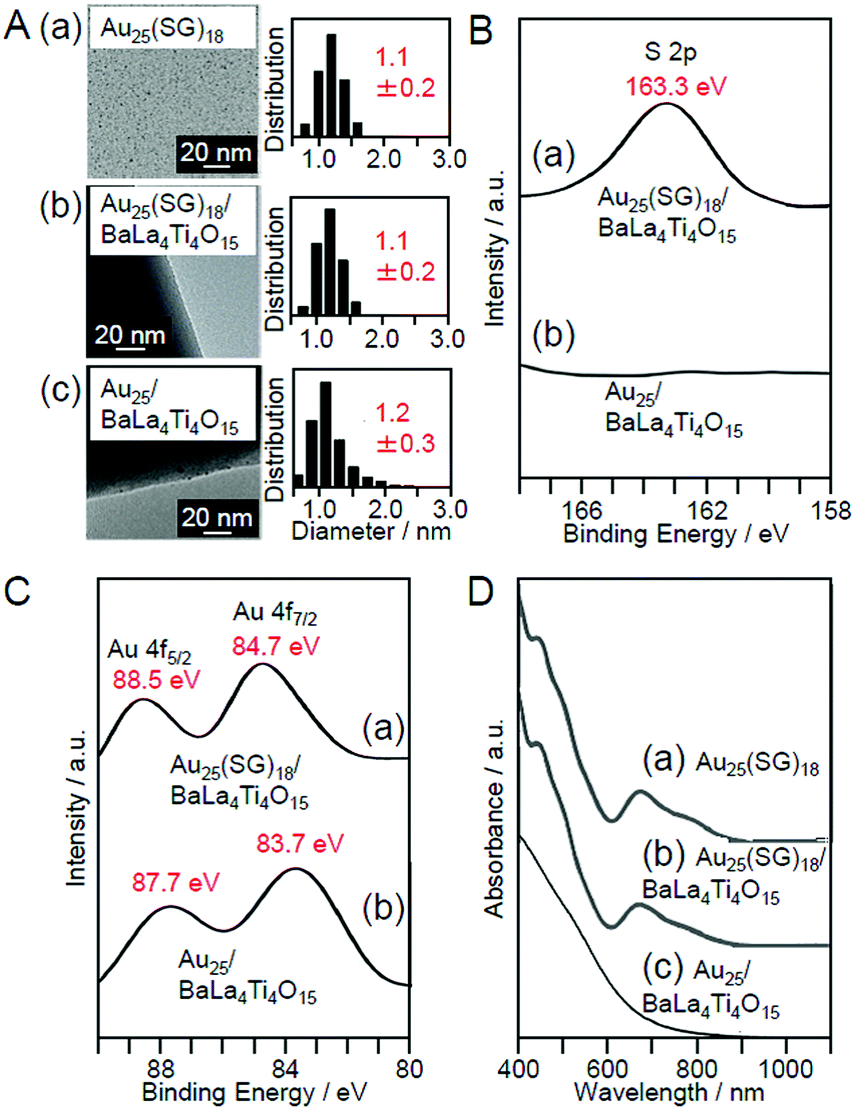 | ||
| Fig. 9 (A) TEM images and size distributions of (a) Au25(SG)18, (b) Au25(SG)18/BaLa4Ti4O15, and (c) Au25/BaLa4Ti4O15 (0.1 wt% Au). (B) S 2p XPS spectra and (C) Au 4f XPS spectra of (a) Au25(SG)18/BaLa4Ti4O15 and (b) Au25/BaLa4Ti4O15. (D) (a) Optical absorption spectra of Au25(SG)18 aqueous solution and the diffuse reflectance spectrum of (b) Au25(SG)18/BaLa4Ti4O15 and (c) Au25/BaLa4Ti4O15. Reproduced with permission from ref. 225. Copyright 2013 Royal Society of Chemistry. | ||
Effect of the types of precursor NCs. We also attempted to load Aun NCs (n represents the number of Au atoms) composed of other numbers of constituent atoms on BaLa4Ti4O15. In this experiment, Aun(SG)m NCs (n = 10, 15, 18, 22, 25, 29, 33, or 39; m represents the number of ligands79) were used as a precursor. First, an aqueous solution containing Aun(SG)m NCs (0.1 wt% Au) and BaLa4Ti4O15 was stirred at room temperature for 1 h. Then, the adsorption efficiency of the Aun(SG)m NCs was estimated by evaluating the amount of Au contained in the supernatant solution after stirring using inductively coupled plasma-mass spectrometry. The results indicated that the Aun(SG)m NCs adsorbed on the photocatalyst with adsorption efficiencies of 98.8% (n = 10), 97.8% (n = 15), 96.6% (n = 18), 99.1% (n = 22), 99.2% (n = 25), 97.9% (n = 29), 96.7% (n = 33), and 99.4% (n = 39), respectively.232 These results indicate that Aun(SG)m NCs with 0.1 wt% Au can be adsorbed on BaLa4Ti4O15 at high adsorption efficiencies regardless of the chemical compositions. However, these results also imply that Aun(SG)m NCs cannot necessarily be adsorbed on BaLa4Ti4O15 with an adsorption efficiency of 100%. The adsorption efficiencies did not exceed the above values even when the stirring time was extended to 2 h. Thus, the results indicate that a chemical equilibrium was achieved between the aqueous solution of the Aun(SG)m NCs and Au25(SG)18/BaLa4Ti4O15 at the above adsorption efficiencies. These results warn that the actual amount of Aun(SG)m NCs adsorbed on BaLa4Ti4O15 cannot be accurately estimated solely from the mixing ratio of Aun(SG)m NCs and BaLa4Ti4O15.
Fig. 10A–C present TEM images and the size distributions of the Aun(SG)m NCs, Aun(SG)m/BaLa4Ti4O15, and Aun/BaLa4Ti4O15, respectively. For all the Aun(SG)m NCs, the average particle sizes and particle-size distributions were relatively similar for the Aun(SG)m NCs and Aun(SG)m/BaLa4Ti4O15 (Fig. 10A and B).232 This finding indicates that no significant change in the particle size (especially the increase of the particle size) of the Aun(SG)m NCs occurs between before and after adsorption. In contrast, different behaviors were observed for Aun/BaLa4Ti4O15 depending on the type of Aun(SG)m NCs. According to Fig. 10, the NCs can be broadly classified into the following two groups based on their similar phenomena: (1) n = 10, 15, 18, 25, and 39 and (2) n = 22, 29, and 33. The Aun NCs in group 1 maintained a narrow distribution of their particle sizes even after calcination, although the average particle size slightly increased during calcination (Fig. 10B and C). In contrast, for the Aun NCs in group 2, the average particle size significantly increased during calcination. These two groups are in good agreement with the classification of the stability of Aun(SG)m NCs against dissociation in aqueous solution: the Aun(SG)m NCs in group 2 dissociate in aqueous solution (release of SG or an Au–SR complex) more quickly than those in group 1.79 From these results, it can be considered that the stability of Aun(SG)m NCs against dissociation is greatly related to the increase in the particle size of the Aun NCs in group 2. Most likely, part of the Aun(SG)m NCs was dissociated during the stirring process, and the Au–SG complex produced by such dissociation was adsorbed on BaLa4Ti4O15 together with the Aun(SG)m NCs. It is presumed that the observed phenomenon occurred because such an Au–SG complex promoted the aggregation of Aun NCs on BaLa4Ti4O15 during calcination. These results demonstrate that using Aun NCs with high stability in solution as a precursor is essential to precisely load the Aun NCs on BaLa4Ti4O15 (Fig. 11).
 | ||
| Fig. 10 TEM images and size distributions of the different (A) Aun(SG)m NCs, (B) Aun(SG)m/BaLa4Ti4O15, and (C) Aun/BaLa4Ti4O15 (0.1 wt% Au). The Aun NCs highlighted in red maintained their particle size during a series of loading processes. Reproduced with permission from ref. 232. Copyright 2015 American Chemical Society. | ||
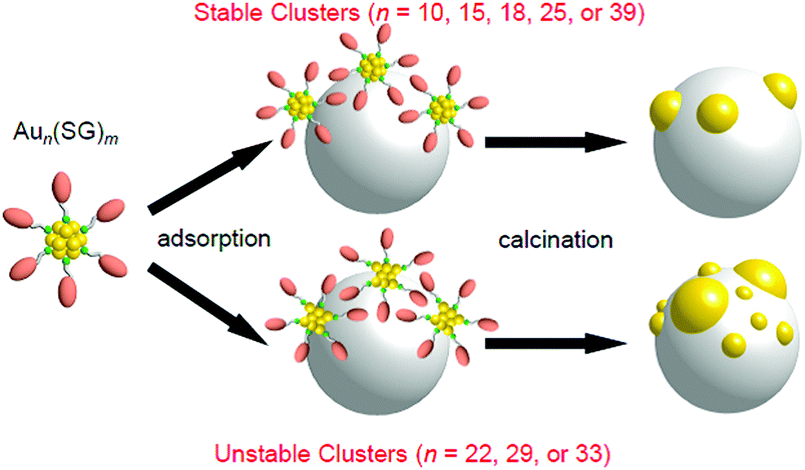 | ||
| Fig. 11 Schematic of the aggregation of Aun(SG)m on the photocatalyst depending on the cluster size (0.1 wt% Au). Reproduced with permission from ref. 232. Copyright 2015 American Chemical Society. | ||
Effect of loading amounts. We also investigated the effect of the amount of adsorbed NCs on the particle size of the loaded NCs using Au25(SG)18 as a precursor. Fig. 12 lists the particle size distributions of the loaded particles, which were estimated from TEM images of photocatalysts containing each amount of Au25(SG)18. When the amount of adsorption of Au25(SG)18 was higher than 0.2 wt% Au, the particle size of the loaded Au NCs increased during calcination.225 Under these conditions, Au25(SG)18 should be adsorbed on BaLa4Ti4O15 at a narrow distance. This appears to induce the aggregation of Au25(SG)18 in the calcination process, leading to the increase of the particle sizes. These results indicate that the loaded amount must be less than 0.2 wt% Au to load Au25 NCs on the photocatalyst while maintaining the particle size of the precursor NCs. The maximum water-splitting activity of the resulting photocatalysts was observed when the amount of loaded Au was 0.1 wt%, and the water-splitting activity decreased when it was greater than 0.2 wt% Au.225 The following two factors are considered to be involved in this phenomenon: (1) a decrease in the proportion of surface Au atoms in the cocatalyst with an increase in particle size and (2) a decrease in the amount of light absorption of the photocatalyst with an increase in the amount of cocatalyst. Based on these results, the amount of loaded cocatalyst was fixed to 0.1 wt% Au in the subsequent studies using Aun NCs and their related alloy NCs as cocatalysts.
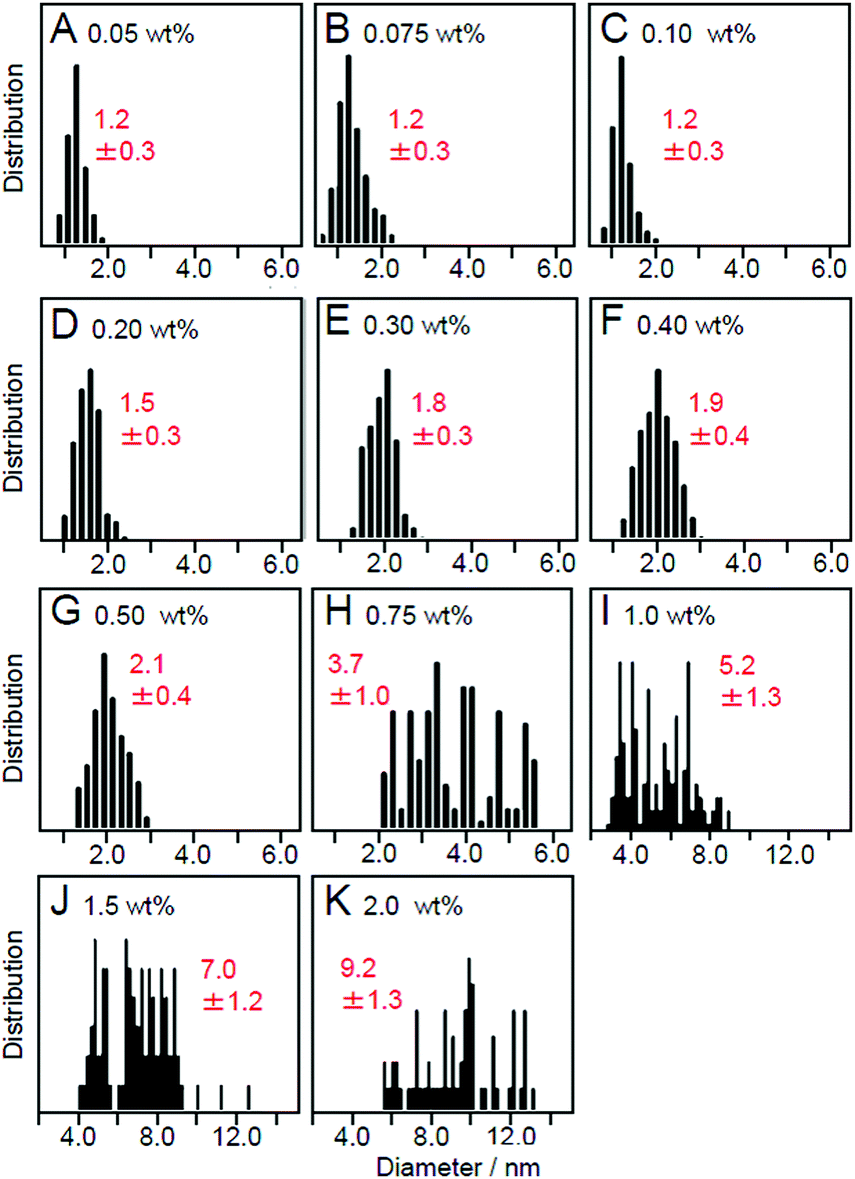 | ||
| Fig. 12 Size distributions of Au NPs estimated from TEM images of Au25/BaLa4Ti4O15 containing (A) 0.05 wt%, (B) 0.075 wt%, (C) 0.10 wt%, (D) 0.20 wt%, (E) 0.30 wt%, (F) 0.40 wt%, (G) 0.50 wt%, (H) 0.75 wt%, (I) 1.0 wt%, (J) 1.5 wt%, and (K) 2.0 wt% Au. Reproduced with permission from ref. 225. Copyright 2013 Royal Society of Chemistry. | ||
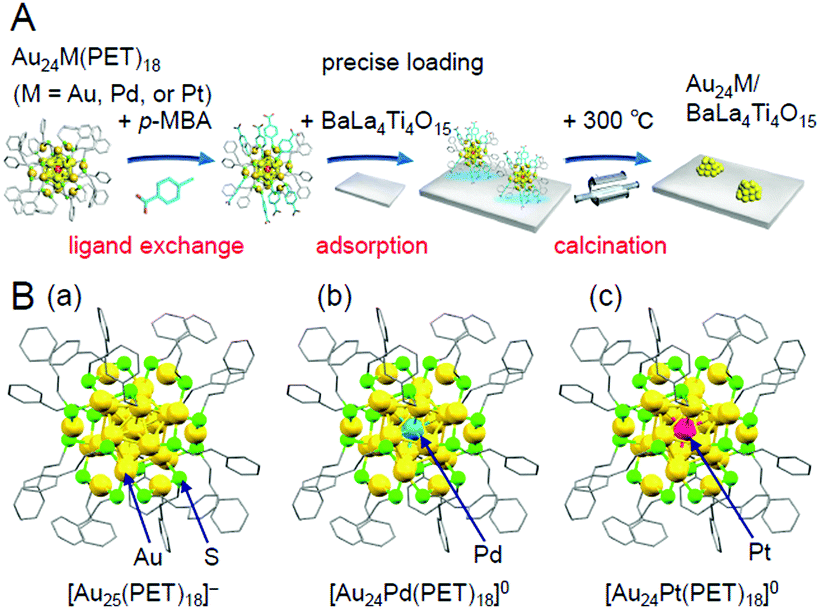 | ||
| Fig. 13 (A) Schematic of the preparation procedure for Au24M/BaLa4Ti4O15 (M = Au, Pd, or Pt) using hydrophobic metal NCs as a precursor.233 (B) Geometric structures of (a) [Au25(PET)18]−,112 (b) [Au24Pd(PET)18]0,96 and (c) [Au24Pt(PET)18]0.96 These metal NCs have been revealed to have a geometric structure where the Au12M metal core (M = Au, Pd, or Pt) is surrounded by six –PET–[Au–PET–]2 staples.96,112 In [Au24Pd(PET)18]0 and [Au24Pt(PET)18]0, both Pd and Pt are located at the central position of the Au12M metal core. Reproduced from ref. 233, 112 and 96. Copyright 2019 American Chemical Society, Copyright 2016 Royal Society of Chemistry, and Copyright 2008 American Chemical Society, respectively. | ||
In this experiment, phenylethanethiolate (PET), which has often been used as a hydrophobic ligand, was used as the ligand. First, Au25(PET)18 (Fig. 13B(a)),112 Au24Pd(PET)18 (Pd = palladium; Fig. 13B(b)),96 and Au24Pt(PET)18 (Fig. 13B(c))96 were synthesized with atomic precision. Then, some of the PET was replaced with hydrophilic p-MBA111 by a ligand-exchange reaction. The matrix-assisted laser desorption/ionization (MALDI) mass spectra obtained for the sample after ligand exchange only contained peaks attributable to Au25(PET)18−y(p-MBA)y (y = 7–14; Fig. 14A), Au24Pd(PET)18−y(p-MBA)y (y = 1–9; Fig. 14B), and Au24Pt(PET)18−y(p-MBA)y (y = 3–10; Fig. 14C). No intrinsic changes were observed in the diffuse reflectance spectra of the NCs or the TEM images before and after ligand exchange. These results indicate that some of the ligands were replaced in each NC without any change in the framework structure of the NC. Each NC was adsorbed on BaLa4Ti4O15 at a high adsorption efficiency of >96% after the ligand exchange (Fig. 14D–F). In the Au L3-edge Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) spectra of the photocatalysts after calcination, almost no peaks attributed to the Au–S bond (∼1.8 Å) were observed (Fig. 14G). The coordination numbers of Au (7.1–7.7) of each of the loaded NCs estimated from the Au L3-edge FT-EXAFS spectra were between those expected for cuboctahedral-structured Au13 (5.5) and Au55 (7.9). In the TEM images of the photocatalysts after calcination, particles with sizes similar to those before calcination were observed. These results confirmed that the ligands were removed from Au25(PET)18−y(p-MBA)y, Au24Pd(PET)18−y(p-MBA)y, and Au24Pt(PET)18−y(p-MBA)y by calcination and, therefore, bare Au25, Au24Pd, and Au24Pt were loaded on BaLa4Ti4O15 without any aggregation.233
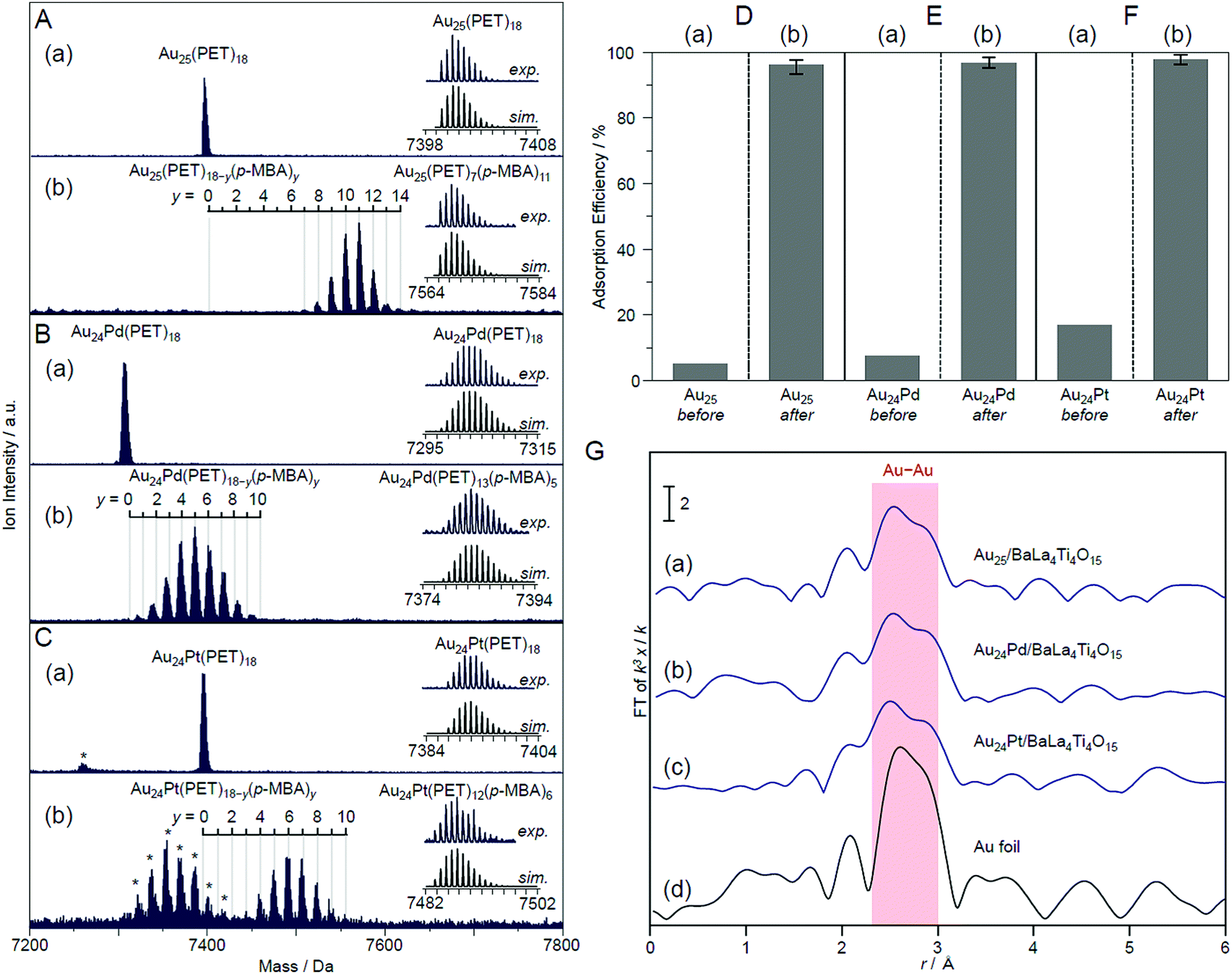 | ||
| Fig. 14 Negative-ion MALDI mass spectra of (a) Au24M(PET)18 and (b) Au24M(PET)18−y(p-MBA)y for M = (A) Au, (B) Pd, and (C) Pt. The experimental and simulated isotope patterns are compared in the inserts. In these spectra, the asterisks indicate the laser fragments. Adsorption efficiency of Au24M(SR)18 for M = (D) Au, (E) Pd, and (F) Pt (SR = PET or p-MBA) on BaLa4Ti4O15 (a) before and (b) after the ligand-exchange reaction. (G) Au L3-edge FT-EXAFS spectra of Au24M/BaLa4Ti4O15 for M = (a) Au, (b) Pd, and (c) Pt together with that of (d) Au foil. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
3.2. Electronic and geometric structures of loaded metal NCs
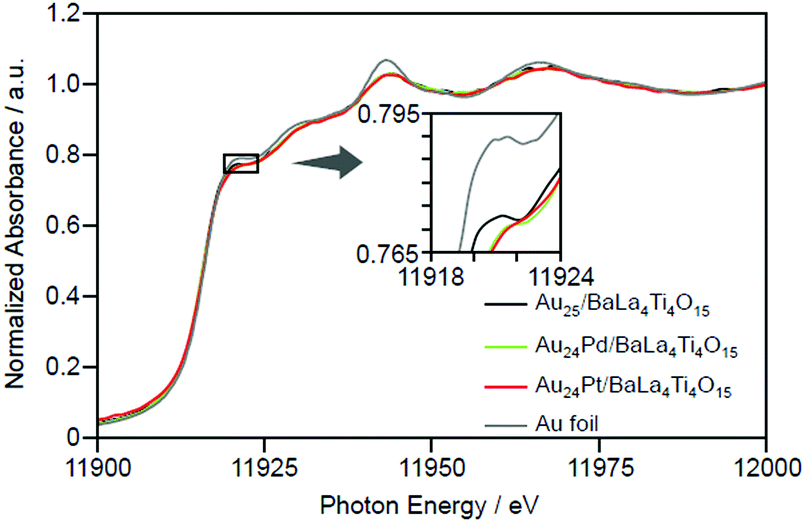 | ||
| Fig. 15 Au L3-edge XANES spectra of Au24M/BaLa4Ti4O15 (M = Au, Pd, and Pt) together with that of Au foil. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
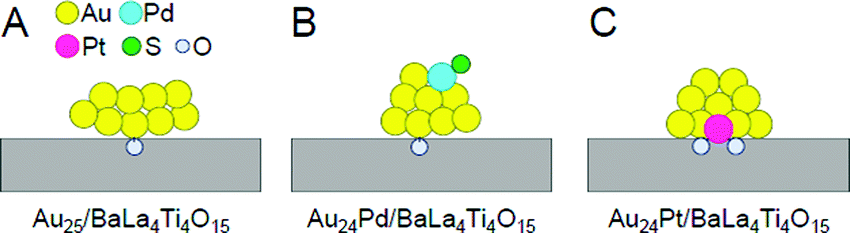 | ||
| Fig. 16 Proposed structures of Au24M/BaLa4Ti4O15 for M = (A) Au, (B) Pd, and (C) Pt. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
Framework structure of Au24M (M = Au, Pd, or Pt). Experiments and DFT calculations by other groups have shown that fine isolated Aun clusters tend to have a planar geometry.234–239 In addition, DFT calculations on Au10/metal oxide predicted that Au10 is most stable on metal oxides when they have a two-dimensional structure.240 Based on these results, Au25 is presumed to have a relatively planar geometry on BaLa4Ti4O15 (Fig. 16A). Indeed, the coordination number of Au estimated from the Au L3-edge FT-EXAFS and the particle size observed in TEM and high-resolution (HR)-TEM images of Au25/BaLa4Ti4O15 strongly support this interpretation.233
On the other hand, Au L3-edge FT-EXAFS, TEM, and HR-TEM measurements suggest that alloy NCs (especially Au24Pt NCs) have a more three-dimensional structure than Au25 NCs.233 For bare Aun NCs produced in the gas phase, it has been shown that doping with Pd or Pt induces the formation of a three-dimensional structure in a smaller size region than for pure Aun.241,242 Au24Pd and Au24Pt can be considered to have a more steric structure than Au25 because a similar heteroatom substitution effect is induced in Au24Pd/BaLa4Ti4O15 and Au24Pt/BaLa4Ti4O15 (Fig. 16B and C).
Substitution position of heteroatoms in Au24M/BaLa4Ti4O15 (M = Pd or Pt). For Au24Pd/BaLa4Ti4O15, the EXAFS results indicate that Pd is located on the surface of the loaded metal NCs and is bound to S (Fig. 16B).233 In the precursor Au24Pd(SR)18, Pd was located at the center of the metal core and was not bound to S (Fig. 13B(b)). Therefore, it is inferred that this geometric structure was formed because Pd moved to the surface of the particles during calcination and combined with S that was separated from Au. On the other hand, Pt was found to be located at the interface between Au24Pt and BaLa4Ti4O15 and bound to several O in BaLa4Ti4O15 (Fig. 16C).233
Pt forms a stronger bond with O than Au (318.4 ± 6.7 kJ mol−1 for Pt–O vs. 223 ± 21.1 kJ mol−1 for Au–O)243 and can bind with multiple O, thereby causing Pt to be located at the interface between Au24Pt NCs and BaLa4Ti4O15 (Fig. 16C). On the other hand, Pd forms a weaker bond with O than Au (145 ± 11.1 kJ mol−1 for Pd–O vs. 223 ± 21.1 kJ mol−1 for Au–O).243 Thus, it is thermodynamically undesirable for Pd to be located at the interface. Because Pd has a higher surface energy than Au (2.003 J m−2 for the Pd (111) surface vs. 1.506 J m−2 for the Au (111) surface),244 it is also thermodynamically undesirable for Pd to be located on the surface. Accordingly, the Pd–S bond appears to be formed to suppress the instability of the NCs (Fig. 16B).
Immobilization of metal NCs at the interface in Au24M/BaLa4Ti4O15 (M = Au, Pd, or Pt). As mentioned above, Au was located at the interface in Au25/BaLa4Ti4O15 and Au24Pd/BaLa4Ti4O15. The binding energy of Au and O is not very high. Moreover, for Au10/metal oxide, DFT calculations predicted that planar Au10 forms an Au–O bond with the metal-oxide surface at a single point.240 In the Au L3-edge FT-EXAFS spectra of Au25/BaLa4Ti4O15 and Au24Pd/BaLa4Ti4O15, no strong peaks attributed to Au–O were observed.233 Thus, it appears that there are only a small number of Au–O bonds in Au25/BaLa4Ti4O15 and Au24Pd/BaLa4Ti4O15, and thus the metal NCs are weakly immobilized on BaLa4Ti4O15 in these photocatalysts (Fig. 16A and B).
On the other hand, in Au24Pt/BaLa4Ti4O15, Pt was located at the interface and formed multiple Pt–O bonds with BaLa4Ti4O15 (Fig. 16C). Pt can bind more strongly with O than Au. Thus, Au24Pt is considered to be more strongly immobilized on BaLa4Ti4O15 than Au25 and Au24Pd. In fact, Au24Pt did not aggregate on BaLa4Ti4O15 as much as Au25 and Au24Pd during the water-splitting reaction.233 This fact also strongly supports our above interpretation.
3.3. Effect of controlling the cocatalysts on the catalytic activity
The effect of the chemical composition of the cocatalyst on the water-splitting activity was successfully elucidated by controlling the chemical composition of the metal NCs with atomic precision. | ||
| Fig. 17 Schematic of the system used to estimate the photocatalytic activity in our study.225,232 Reproduced with permission from ref. 247. Copyright 2018 American Chemical Society. | ||
Under conditions of flowing CO2. In our early studies,225,232 CO2 was flowed into the reaction cell together with argon (Ar) to accelerate the water-splitting reaction.246 In these experiments, the evolution of H2 and O2 increased continuously over time. The volume ratio of the evolved gas was H2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 = 2:1. These results indicated that the water-splitting photocatalysis proceeded ideally under the conditions of this gas flow.
:O2 = 2:1. These results indicated that the water-splitting photocatalysis proceeded ideally under the conditions of this gas flow.
Fig. 18 shows the rate of gas evolution over Aun/BaLa4Ti4O15 (n = 10, 15, 18, 25, and 39; 0.1 wt% Au) and AuNP/BaLa4Ti4O15 (AuNP = Au NPs) on which 8–22 nm of AuNP was loaded by photodeposition. As shown in Fig. 18, the amount of gas increased continuously with a decreasing number of constituent atoms.232 This finding indicates that Aun/BaLa4Ti4O15 loaded with smaller sized Aun NCs has higher photocatalytic activity for Aun/BaLa4Ti4O15 with the same amount (wt%) of Au. Because the increase in activity with a decreasing number of constituent atoms is very gradual, the change in activity between them is interpreted to be mainly caused by the change in the ratio of surface Au atoms, i.e., the change in the number of Au atoms that can react with protons (H+).
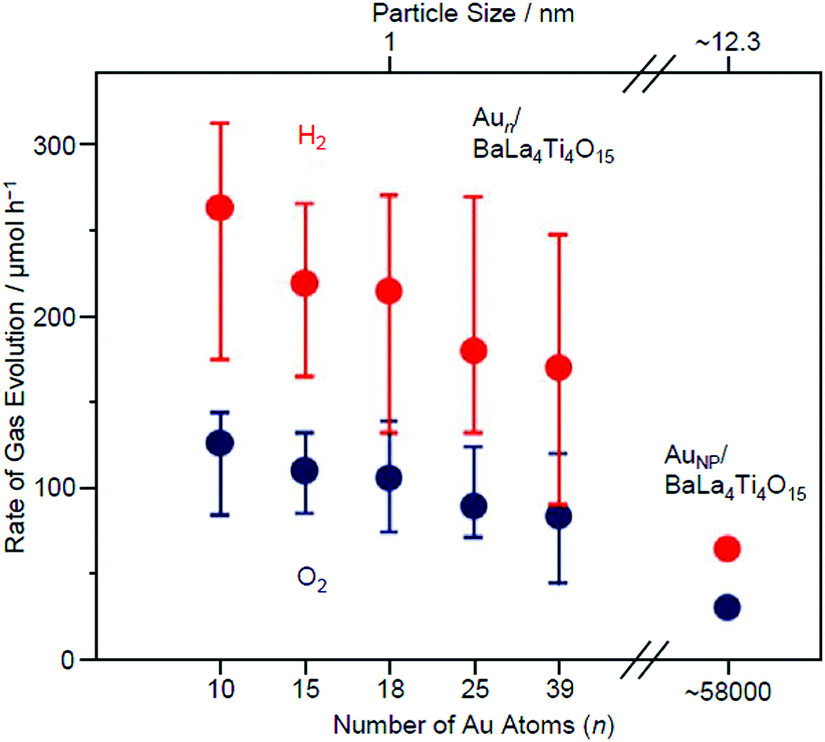 | ||
| Fig. 18 Effect of the NC size on the water-splitting activity studied using Aun/BaLa4Ti4O15 (n = 10, 15, 18, 25, and 39) and AuNP/BaLa4Ti4O15 (0.1 wt% Au). The average values obtained from four measurements are plotted herein. Reproduced with permission from ref. 232. Copyright 2015 American Chemical Society. | ||
On the other hand, the difference in photocatalytic activity between Aun/BaLa4Ti4O15 and AuNP/BaLa4Ti4O15 cannot be explained by the difference in the number of surface atoms alone. Various analyses have shown that the activity per surface Au atom was reduced by 75–85% in Au10/BaLa4Ti4O15 compared with in AuNP/BaLa4Ti4O15.232 This finding suggests that the increase in activity by the ultra-miniaturization of the Au cocatalyst (Au NPs → Aun NCs) (Fig. 18) is mainly caused by the increase in the number of surface Au atoms, which exceeds the decrease in activity per Au atom.
Without flowing CO2. Although the CO2 flow in the above-mentioned experiments is an effective means to achieve high activity, the photocatalysts are expected to be dispersed over a large area of water and irradiated with sunlight in practical applications.38 Therefore, we conducted the same measurements under Ar flow conditions (Fig. 17).247 The results revealed that the presence of CO2 affects not only the water-splitting activity but also its dependence on the size of the Au cocatalyst.
Fig. 19A and B show the rate of gas evolution for Au25/BaLa4Ti4O15 and AuNP/BaLa4Ti4O15 under an Ar flow (without CO2). The ratio of H2 to O2 evolution was close to 2:1 regardless of the type of photocatalyst (Au25/BaLa4Ti4O15 or AuNP/BaLa4Ti4O15), confirming that the water-splitting reaction proceeded ideally. However, under this flow condition, there was no significant difference in the water-splitting activity between Au25/BaLa4Ti4O15 and AuNP/BaLa4Ti4O15. This finding indicates that the size effect of the cocatalyst varies depending on the type of flowing gas and that the water-splitting activity cannot be improved by mere miniaturization of the Au cocatalyst in the absence of a CO2 flow.
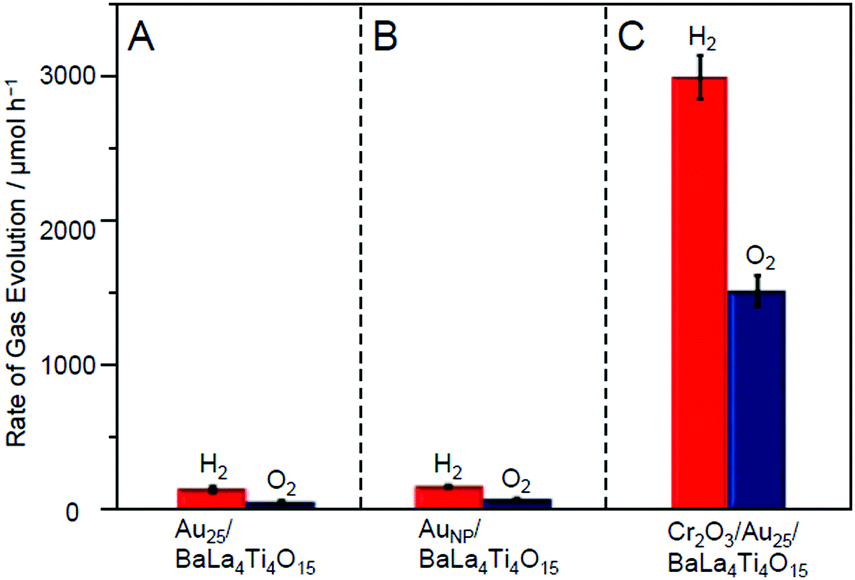 | ||
| Fig. 19 Comparison of the rates of H2 and O2 evolution by photocatalytic water-splitting over (A) Au25/BaLa4Ti4O15, (B) AuNP/BaLa4Ti4O15, and (C) Cr2O3/Au25/BaLa4Ti4O15 (0.1 wt% Au; 0.5 wt% Cr). Averages of values obtained from several experiments are shown. Reproduced with permission from ref. 247. Copyright 2018 American Chemical Society. | ||
The water-splitting reaction consists of multiple reactions (Fig. 20). Therefore, we investigated how the miniaturization of the Au cocatalyst affects each reaction. First, we examined the HER (Fig. 20A). The results revealed that the miniaturization of Au cocatalysts accelerate the HER. However, as mentioned above, the water-splitting activity was not improved by miniaturization of the Au cocatalyst. Thus, we next investigated the effect of the miniaturization of Au cocatalysts on the reverse reaction. The results indicated that the miniaturization of the Au cocatalyst also accelerates one of the reverse reactions, the O2-photoreduction reaction (Fig. 20D). It was thus interpreted that under an Ar flow, the water-splitting activity cannot be enhanced by mere miniaturization of the Au cocatalyst as it causes the acceleration of both reactions, thereby cancelling the acceleration effect.247
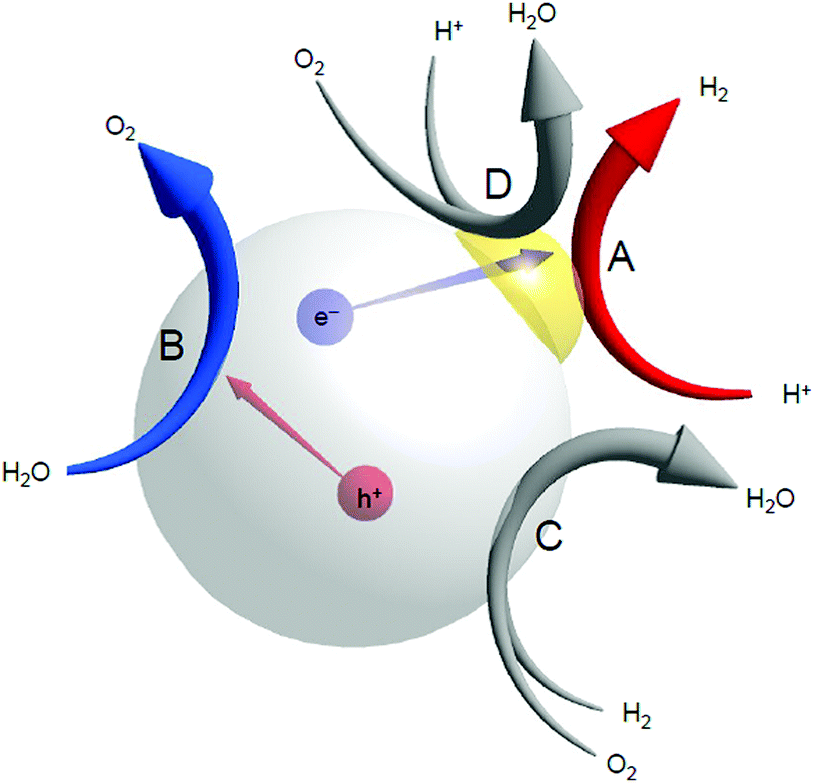 | ||
| Fig. 20 Possible reactions that occur over a water-splitting photocatalyst during the photocatalytic reaction: (A) the HER, (B) the OER, (C) the reverse reaction, and (D) O2 photoreduction. | ||
Fig. 21 shows the amounts of H2 and O2 evolved over Au25/BaLa4Ti4O15, Au24Pd/BaLa4Ti4O15, and Au24Pt/BaLa4Ti4O15. The amount of evolved gas was higher in the order of Au24Pd/BaLa4Ti4O15 < Au25/BaLa4Ti4O15 < Au24Pt/BaLa4Ti4O15. This result demonstrates that the substitution of one Au atom of Au25/BaLa4Ti4O15 with Pd causes a decrease in the water-splitting activity, whereas the substitution of one Au atom of Au25/BaLa4Ti4O15 with Pt induces an increase in the water-splitting activity. Such an enhancement of the water-splitting activity by Pt substitution has also been observed in the work of another group,248–250 where larger Au–Pt alloy NPs were used as cocatalysts. Fig. 21 demonstrates that such an effect occurs with only single atom substitution of the cocatalyst for Au25/BaLa4Ti4O15.
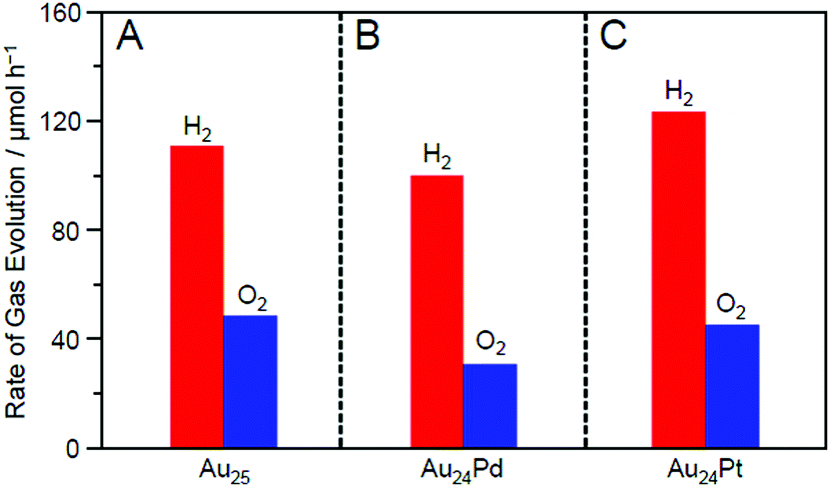 | ||
| Fig. 21 Rates of H2 and O2 evolution by photocatalytic water-splitting with Au24M/BaLa4Ti4O15 for M = (A) Au, (B) Pd, and (C) Pt (∼0.1 wt% M; all the samples include the same number of metal atoms in the cocatalysts). The averages of the values obtained from three experiments are used in the figures. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
To elucidate the reason for this change in the water-splitting activity, we investigated the effect of heteroatom substitutions on each reaction over the photocatalyst (Fig. 20).233 The results indicated that both heteroatom substitutions accelerated the H2 evolution (Fig. 22). These substitutions were also observed to accelerate the O2 photoreduction, especially for the Pd substitution. From these results, it can be interpreted that the water-splitting activity was reduced by Pd substitution because its effect on the acceleration of O2 photoreduction exceeded its effect on the enhancement of H2 evolution, whereas the water-splitting activity was increased by Pt substitution because the effect of improving H2 evolution exceeded the effect of promoting O2 photoreduction (Fig. 22).
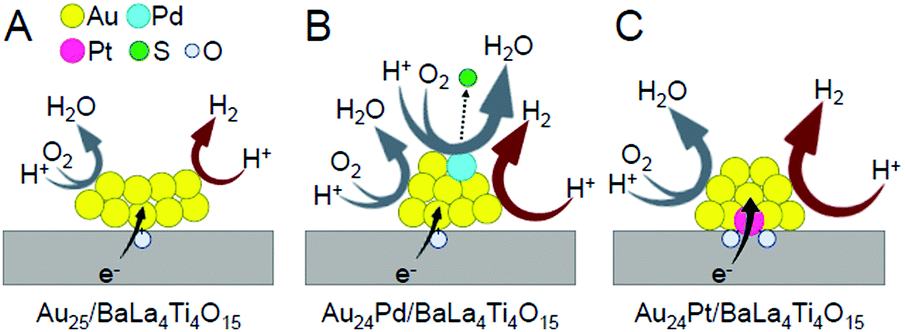 | ||
| Fig. 22 Proposed structures of Au24M/BaLa4Ti4O15 for M = (A) Au, (B) Pd, and (C) Pt during the water-splitting reaction. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
Thus, the Pd and Pt substitutions induce different effects on the reactions that occur on the photocatalyst, respectively. This appears to be largely related to the different substitution positions of both elements. As mentioned above, both Pd and Pt substitutions increase the electron density of Au (Fig. 15). Because both H2 production and O2 photoreduction require electrons (Fig. 20), this increase in the electron density of Au appears to cause an increase in the reaction rates of both reactions (Fig. 22B and C).
However, in Au24Pd/BaLa4Ti4O15, Pd, which more easily reduces O2 than Au, is located on the surface of the alloy NCs (Fig. 22B). Pd was bound to S immediately after the photocatalyst preparation (Fig. 16B). However, after light irradiation, S appears to be reduced to H2S and then desorbed from the surface of the metal NCs. Thus, Pd is presumed to be exposed during the water-splitting reaction (Fig. 22B). The presence of such bare Pd is expected to greatly accelerate the O2 photoreduction over Au24Pd/BaLa4Ti4O15. This appears to be the reason why the effect of Pd substitution on the acceleration of O2 photoreduction exceeded the effect on the enhancement of H2 evolution.
For Au24Pt/BaLa4Ti4O15, the Pt is located at the interface between the alloy NCs and BaLa4Ti4O15 (Fig. 16C); thus, Pt is not exposed during the water-splitting reaction. In addition, because electron transfer occurs more easily via Pt atoms than via Au atoms at the interface, it is expected that photoexcited electrons are more efficiently transferred from BaLa4Ti4O15 to metal NCs in Au24Pt/BaLa4Ti4O15 than in Au24Pd/BaLa4Ti4O15 (and Au25/BaLa4Ti4O15) (Fig. 22C). These facts appear to be largely related to the difference in the heteroatom substitution effect between Pd and Pt substitutions.
3.4. Creation of highly active water-splitting photocatalysts
In this way, the establishment of the atomically precise control technique of cocatalysts provided a deeper understanding of (i) the electronic/geometric structure of cocatalysts, (ii) the binding mode at the interface between the cocatalysts and photocatalysts, and (iii) the effect of the number of constituent atoms of the cocatalysts and the alloying on the catalytic activities. We have also attempted to create highly active water-splitting photocatalysts based on this knowledge.One effective means to suppress the reverse reaction of water splitting is to form a chromium oxide (Cr2O3) shell on the surface of the loaded cocatalyst. The Cr2O3 shell is permeable to H+ but not to O2 approaching from the outside. Domen et al. reported that when the cocatalyst surface is covered by a Cr2O3 shell with such characteristics, it is possible to suppress only the progress of the reverse reaction while maintaining the H2-evolution activity. Thus, this approach is effective for improving the water-splitting activity.38,146,219–221,251–256 In their research, they used the photodeposition method to form the Cr2O3 shell (Fig. 23A). However, when Au25/BaLa4Ti4O15, Au24Pd/BaLa4Ti4O15, and Au24Pt/BaLa4Ti4O15 were irradiated with UV light, the loaded metal NCs aggregated.233 Thus, when the method reported by Domen et al. is used as is, it is difficult to form the Cr2O3 shell on the surface of the loaded metal NCs while maintaining the number of atoms of the metal NCs. However, research in the field of surface science has revealed that when a metal oxide loaded with metal NPs is heated under an H2 or O2 atmosphere, a strong metal–support interaction (SMSI) is induced, thereby leading to the formation of an oxide film on the metal NPs.257–264 Therefore, we attempted to form a Cr2O3 shell on the surface of the metal NC cocatalyst using such a SMSI effect (Fig. 23B).247
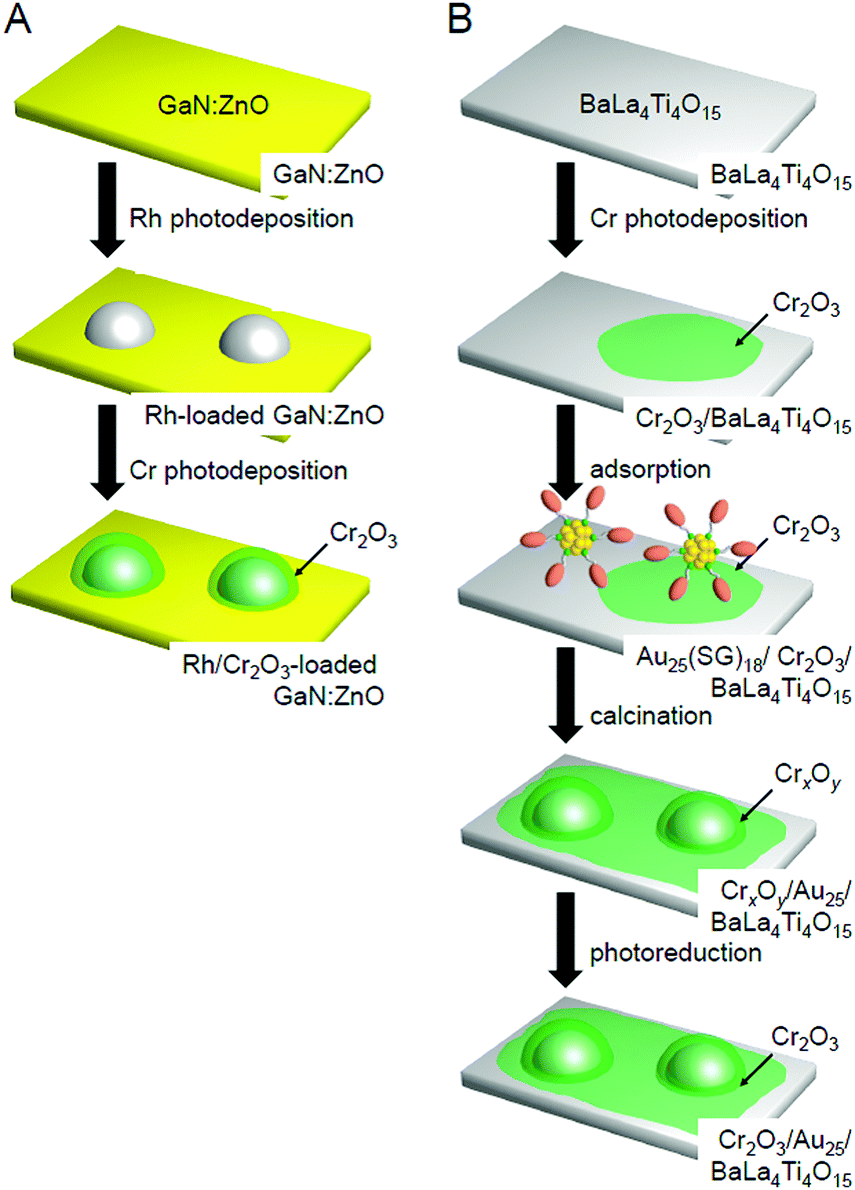 | ||
| Fig. 23 Comparison of the procedures of Cr2O3 shell formation in (A) the literature254 and (B) our work.247 Reproduced with permission from ref. 247 and 254. Copyright 2018 American Chemical Society and Copyright 2006 Wiley-VCH. | ||
Au25/BaLa4Ti4O15. In our Cr2O3 shell formation process, the Cr2O3 layer was first formed on BaLa4Ti4O15 using the photodeposition method before loading Au25. Fig. 24A and B present TEM and HR-TEM images of the photocatalysts on which the Cr2O3 (0.5 wt% Cr) layer was formed. These images confirm that Cr2O3 layers with thicknesses of approximately 0.7–1.3 nm were formed on BaLa4Ti4O15. Then, Au25(SG)18 was adsorbed onto the obtained Cr2O3/BaLa4Ti4O15 (0.1 wt% Au; Fig. 24C and D). The photocatalyst after the adsorption of Au25(SG)18 (Au25(SG)18/Cr2O3/BaLa4Ti4O15) was calcined at 300 °C for 2 h under a reduced pressure. In the TEM image of the photocatalyst after calcination (Fig. 24E), particles with an average size of 1.1 ± 0.3 nm were observed. In the HR-TEM image of the photocatalyst after calcination (Fig. 24F), a thin layer with a thickness of approximately 0.7–0.9 nm was observed around particles with high electron density. This indicates that the Au25 was covered with a Cr2O3 layer during calcination (Fig. 23B). Part of the chromium was oxidized to a highly oxidized state (>3+) during calcination. Accordingly, we irradiated the photocatalyst with UV light to reduce the highly oxidized chromium oxide to Cr2O3 and thereby obtain the desired Cr2O3/Au25/BaLa4Ti4O15.247
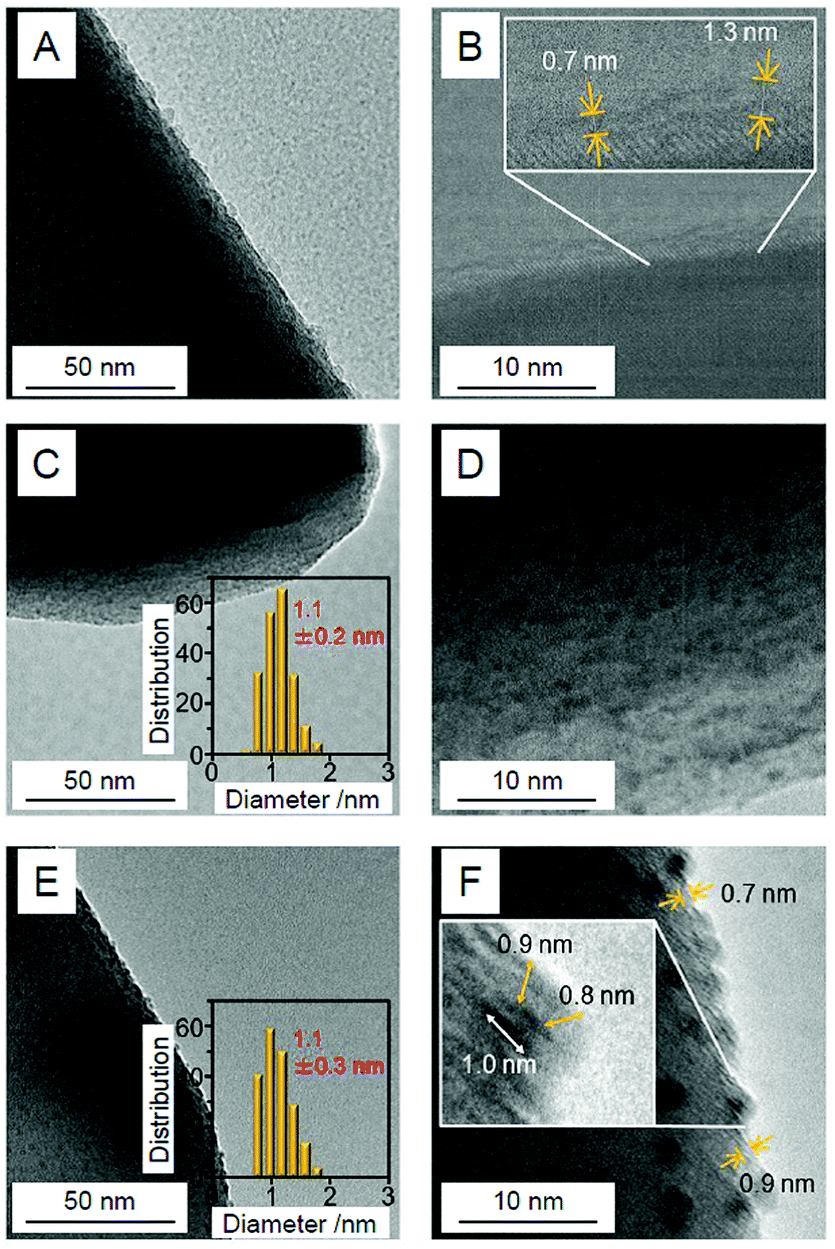 | ||
| Fig. 24 TEM images (A, C and E) and HR-TEM images (B, D and F) of (A and B) Cr2O3/BaLa4Ti4O15 (0.5 wt% Cr), (C and D) Au25(SG)18/Cr2O3/BaLa4Ti4O15 (0.1 wt% Au; 0.5 wt% Cr), and (E and F) Cr2Oy/Au25/BaLa4Ti4O15 (0.1 wt% Au; 0.5 wt% Cr). Cr2Oy indicates the chromium oxide in which part of the chromium was oxidized to a highly oxidized state (>3+). In (C and E), the insets show the core-size distributions of the particles. In (B and F), the thicknesses of the Cr2O3 or Cr2Oy shells are indicated by yellow double-pointed arrows. In part (F), the particle size is also indicated by a white double-pointed arrow. Reproduced with permission from ref. 247. Copyright 2018 American Chemical Society. | ||
The resulting Cr2O3/Au25/BaLa4Ti4O15 exhibited higher water-splitting activity than Au25/BaLa4Ti4O15. Fig. 19A and C show the water-splitting activity of Au25/BaLa4Ti4O15 and Cr2O3/Au25/BaLa4Ti4O15. Au25/BaLa4Ti4O15, without a Cr2O3 shell, evolved H2 at 160.5 μmol h−1 (Fig. 19A). On the other hand, Cr2O3/Au25/BaLa4Ti4O15, which had a Cr2O3 shell, evolved H2 at 3032 μmol h−1 (Fig. 19C). These results demonstrate that forming the Cr2O3 shell enhanced the water-splitting activity of the photocatalyst by approximately 19 times. Cr2O3 itself hardly improved the water-splitting activity.247 In addition, forming the Cr2O3 shell had little effect on the electronic state of Au25.247 Therefore, it can be considered that the improvement in the water-splitting activity was mainly caused by the suppression of the O2-photoreduction reaction due to the Cr2O3 shell formation. Indeed, it was experimentally confirmed that the O2-photoreduction reaction was greatly suppressed over Cr2O3/Au25/BaLa4Ti4O15.247 In this way, we succeeded in creating a highly active water-splitting photocatalyst that takes advantage of Au25 cocatalysts by forming a Cr2O3 shell on the surface of the Au25 cocatalysts.
This study also revealed that the aggregation of cocatalysts during light irradiation can be suppressed by forming the Cr2O3 shell.247 When using our Cr2O3-shell formation method, Au25 was covered with a Cr2O3 layer on BaLa4Ti4O15 (Fig. 23B). In this case, Au25 should not easily move around on the surface of the photocatalysts. Indeed, Cr2O3/Au25/BaLa4Ti4O15 maintained high water-splitting activity for a long time (Fig. 25A). Unlike for Au25/BaLa4Ti4O15 (Fig. 25B(a)), the particle size of the Au cocatalyst hardly changed even after 10 h light-irradiation for Cr2O3/Au25/BaLa4Ti4O15 (Fig. 25B(b)). These results indicate that the Cr2O3 shell formed using our method improves not only the water-splitting activity but also the stability of the cocatalyst on the photocatalyst surface.
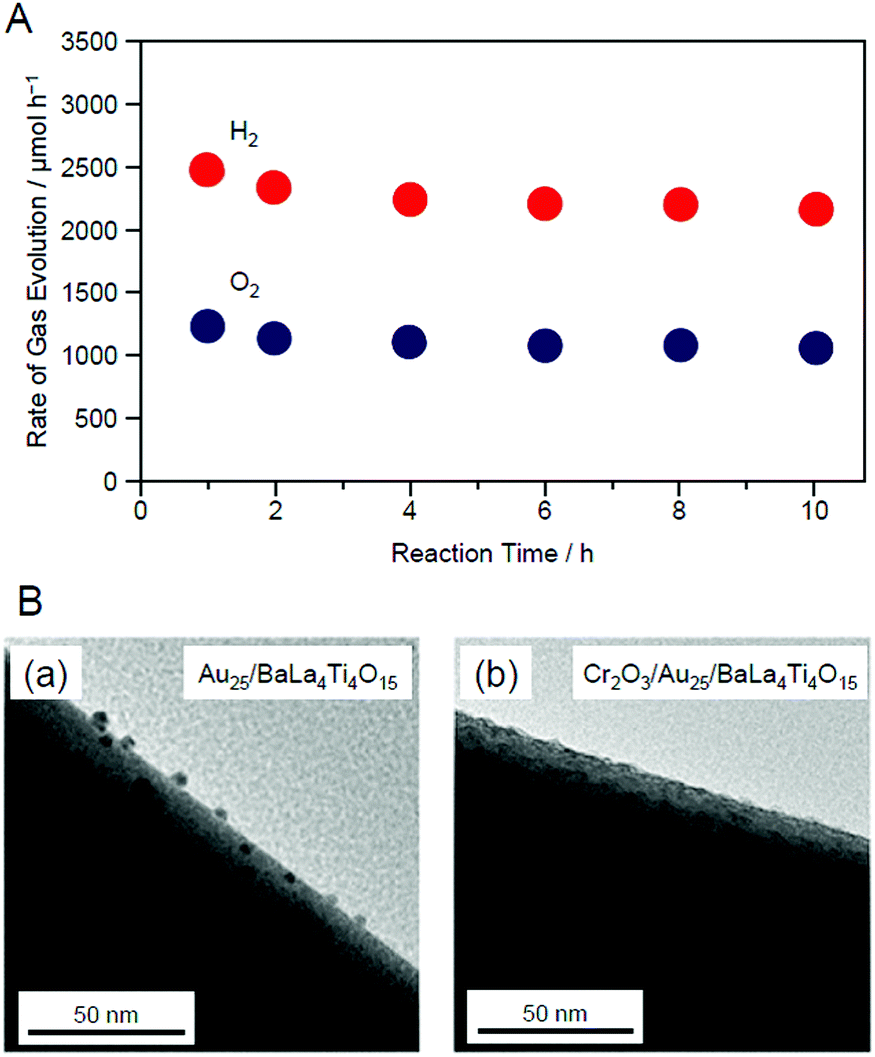 | ||
| Fig. 25 (A) Time dependence of the evolution of H2 and O2 over Cr2O3/Au25/BaLa4Ti4O15 with 0.1 wt% Au and 0.5 wt% Cr. (B) TEM images of (a) Au25/BaLa4Ti4O15 and (b) Cr2O3/Au25/BaLa4Ti4O15 after UV irradiation for 10 h. Reproduced with permission from ref. 247. Copyright 2018 American Chemical Society. | ||
Au24Pt/BaLa4Ti4O15. We also attempted to form the Cr2O3 shell on Au24Pt/BaLa4Ti4O15 using a similar method to that shown in Fig. 23B.233Fig. 26A presents TEM and HR-TEM images of the obtained photocatalyst, respectively. In the TEM image, particles of 1.3 ± 0.3 nm in size were observed. This particle size was slightly larger than that of Au24Pt(PET)18−y(p-MBA)y/Cr2O3/BaLa4Ti4O15 (1.0 ± 0.2 nm). This indicates that for Au24Pt/BaLa4Ti4O15, slight aggregation of the cocatalysts occurred during Cr2O3 shell formation. Based on the particle-size distribution (Fig. 26A(a)), the main product was estimated to be (Au24Pt)1−3 composed of 1–3 Au24Pt units, meaning that ∼77% of the Au24Pt was aggregated to form (Au24Pt)2,3.233p-MBA has a smaller number of hydrophilic functional groups than SG. Furthermore, in Au24Pt(PET)18−y(p-MBA)y(y = 3–10), not all the ligands have hydrophilic functional groups. Therefore, Au24Pt(PET)18−y(p-MBA)y (y = 3–10) should more weakly adsorb to the Cr2O3 surface than Au25(SG)18. It can be considered that slight aggregation of Au24Pt occurred during calcination, most likely due to this weaker adsorption. The HR-TEM images confirmed that the Cr2O3 layer was also formed around (Au24Pt)1−3 (Fig. 26A(b)).
 | ||
| Fig. 26 (A and B) (a) TEM images and particle-size distributions and (b) HR-TEM images of Cr2O3/(Au24Pt)1−3/BaLa4Ti4O15 (∼0.1 wt% M (M = Au or Pt); 0.3 wt% Cr) before (A) and after (B) UV light irradiation for 10 h. The thicknesses of the Cr2O3 shells and the particle sizes are indicated by black and yellow double-ended arrows, respectively. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
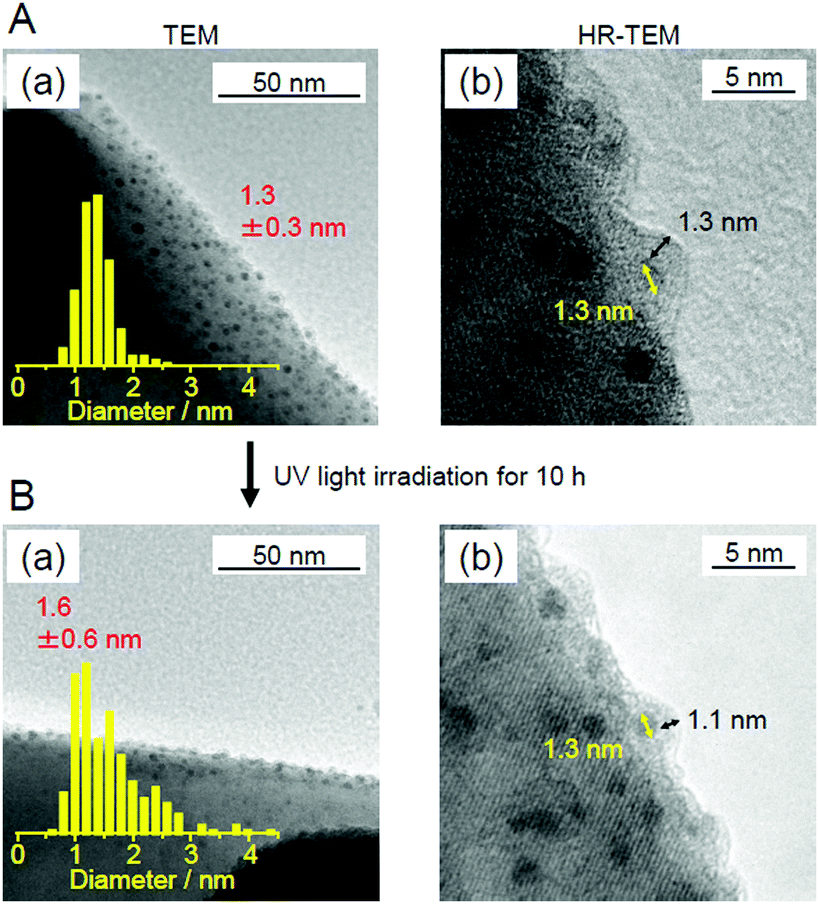
Fig. 27 shows the amount of gas evolved over Cr2O3/(Au24Pt)1−3/BaLa4Ti4O15. This photocatalyst evolved approximately 20 times more gas than Au24Pt/BaLa4Ti4O15 (Fig. 27),233 indicating that Cr2O3/(Au24Pt)1−3/BaLa4Ti4O15 was even more active than Au24Pt/BaLa4Ti4O15. In addition, the total amount of gas increased proportionally with time. After light irradiation, the average particle size slightly increased compared with that of Cr2O3/(Au24Pt)1−3/BaLa4Ti4O15 before light irradiation (Fig. 26B(a)). However, the increase in particle size (1.3 ± 0.3 nm → 1.6 ± 0.6 nm) was far more suppressed than for Au24Pt/BaLa4Ti4O15 without the Cr2O3 layer (1.1 ± 0.2 nm → 2.8 ± 0.9 nm). These results suggest that the combination of Pt substitution and Cr2O3 layer formation makes it possible to create highly active and stable photocatalysts.
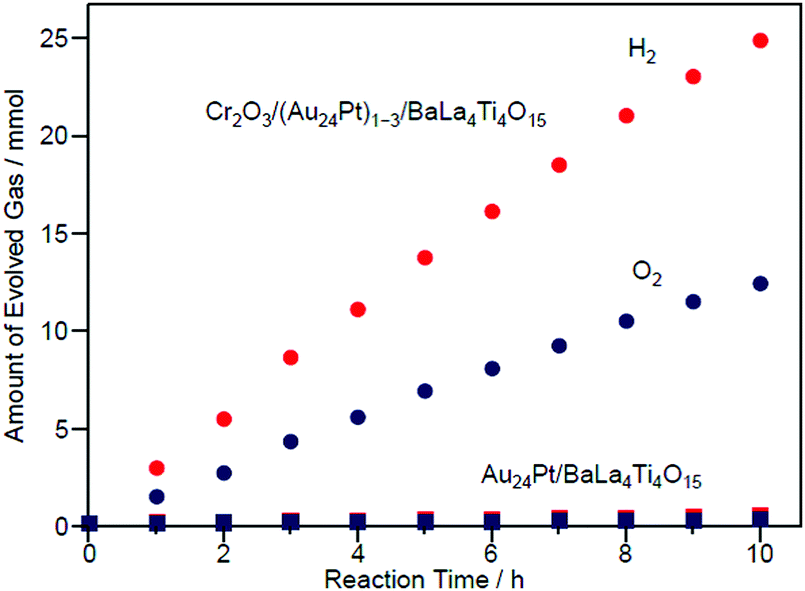 | ||
| Fig. 27 Time course of water splitting over Cr2O3/(Au24Pt)1−3/BaLa4Ti4O15 together with that over Au24Pt/BaLa4Ti4O15. Reproduced with permission from ref. 233. Copyright 2019 American Chemical Society. | ||
In this way, we have succeeded in forming a Cr2O3 layer on (Au24Pt)1−3 and thereby creating a photocatalyst with high activity and stability. Unfortunately, for Au24Pt, ∼77% of the Au24Pt aggregated during formation of the Cr2O3 layer. Furthermore, there was a slight increase in the particle size after 10 h of the water-splitting reaction, even for the photocatalyst with a Cr2O3 layer. This increase is most likely because all of the alloy clusters were not necessarily covered with a Cr2O3 layer. However, if these problems could be addressed, more functional photocatalysts will be successfully created through a combination of decreasing the particle size, alloying, and suppressing the reverse reaction by Cr2O3-layer formation.
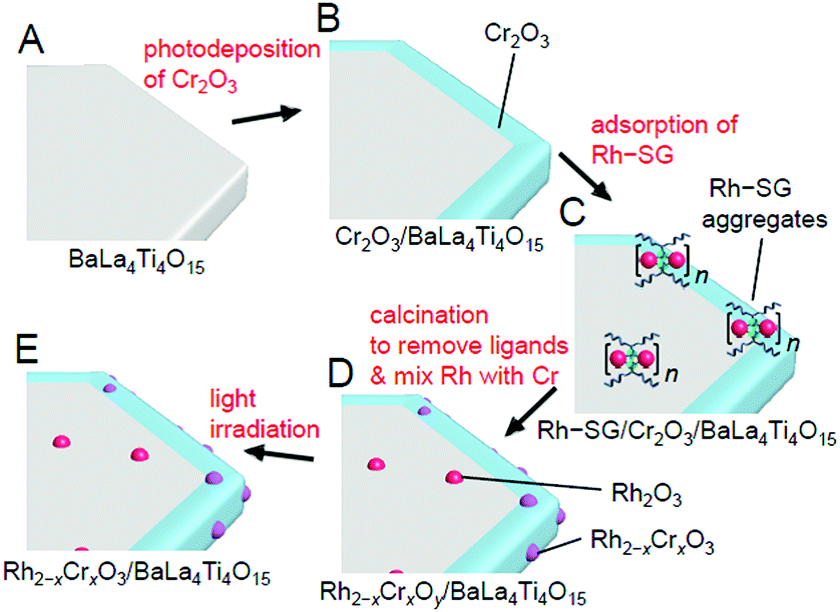 | ||
| Fig. 28 Schematic of the experimental procedure for the formation of Rh2−xCrxO3/BaLa4Ti4O15. (A) BaLa4Ti4O15, (B) Cr2O3/BaLa4Ti4O15, (C) Rh–SG/Cr2O3/BaLa4Ti4O15, (D) Rh2−xCrxOy/BaLa4Ti4O15, and (E) Rh2−xCrxO3/BaLa4Ti4O15. Rh2−xCrxOy indicates Rh2−xCrxO3 including highly oxidized Cr (>3+). Reproduced with permission from ref. 270. Copyright 2020 Wiley-VCH. | ||
Unfortunately, there have been no reports on the precise synthesis of Rh2−xCrxO3 NCs. Therefore, in this experiment, Rh–SG complexes, containing Rh2(SG)2 as a main component, were used as a precursor. First, the Cr2O3 layer was formed on BaLa4Ti4O15 by photodeposition to prepare Cr2O3/BaLa4Ti4O15 (Fig. 29A). Then, the Rh–SG complexes were adsorbed on the surface of Cr2O3/BaLa4Ti4O15 (Fig. 29B(a)). Various structural analyses revealed that approximately 6 Rh2(SG)2 complexes aggregated during adsorption.270 Then, Rh–SG/Cr2O3/BaLa4Ti4O15 was calcined at 300°C under reduced pressure to remove the ligands from the Rh–SG complexes and form a solid solution of Rh and Cr oxides (Fig. 29B and C). Finally, a small amount of Cr with a slightly higher oxidation state was reduced to CrIII by UV light irradiation (Fig. 28E). This series of procedures enabled us to load Rh2−xCrxO3 NCs with sizes of approximately 1.3 ± 0.3 nm and a narrow size distribution on BaLa4Ti4O15 (Rh2−xCrxO3/BaLa4Ti4O15; Fig. 29D(a)).
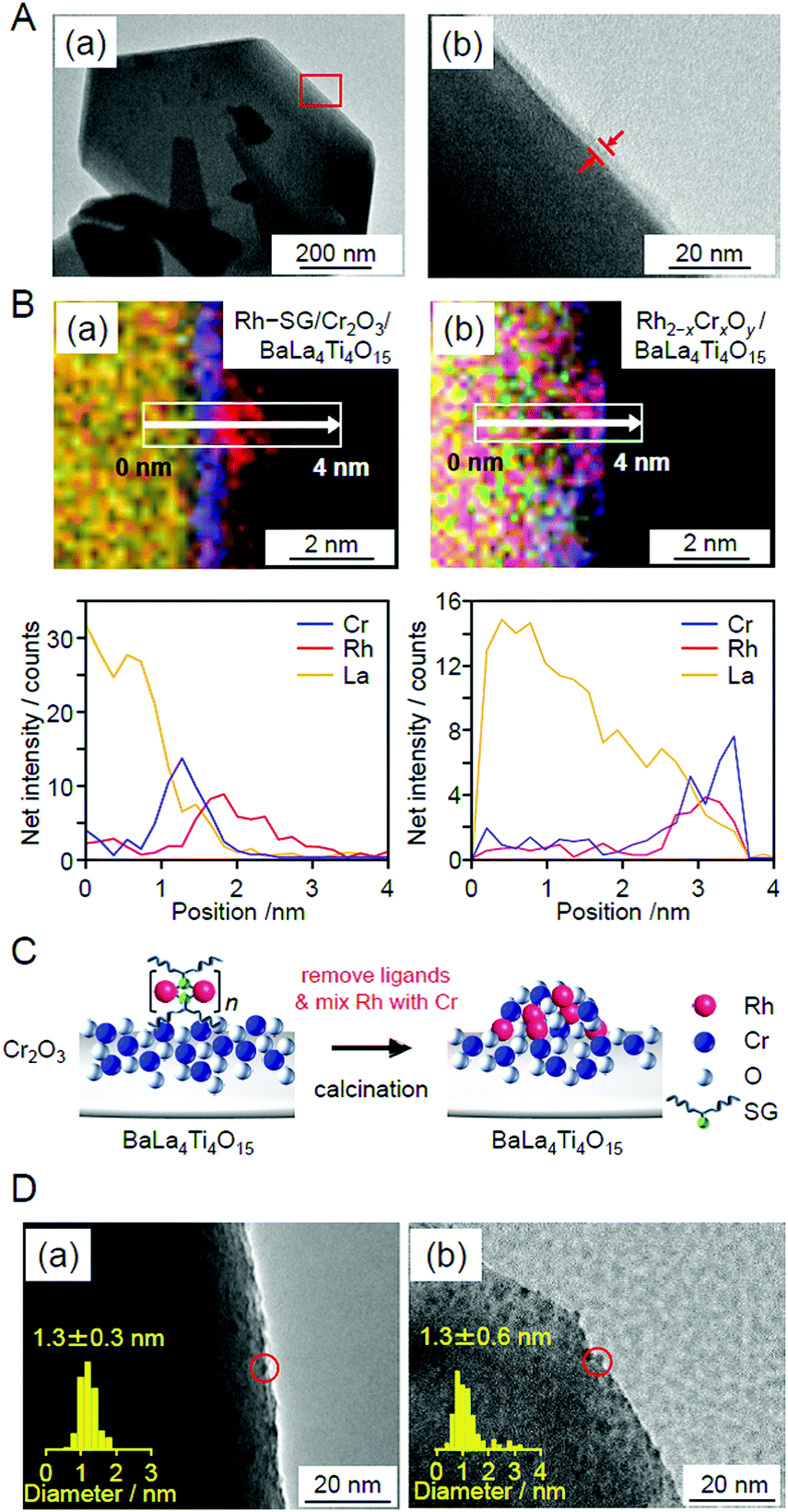 | ||
| Fig. 29 (A) HR-TEM images of Cr2O3/BaLa4Ti4O15 observed at (a) low magnification and (b) high magnification for the edge of BaLa4Ti4O15. The image in (b) is an expansion of the red square in the image in (a). In this experiment, Cr was loaded at 1 wt% to easily monitor the position of the Cr2O3 layers. (B) Line analysis of elemental mapping for (a) Rh–SG/Cr2O3/BaLa4Ti4O15 and (b) Rh2−xCrxOy/BaLa4Ti4O15. (C) Schematic of the phenomenon that occurred during the calcination process. (D) TEM images of Rh2−xCrxO3/BaLa4Ti4O15 (a) before and (b) after UV irradiation for 10 h. The red circles indicate the Rh2−xCrxO3 particles. Reproduced with permission from ref. 270. Copyright 2020 Wiley-VCH. | ||
The obtained photocatalyst exhibited an apparent quantum yield of 16% (excitation wavelength = 270 nm; Fig. 30), which is the highest achieved for BaLa4Ti4O15 to date.270 For this photocatalyst, almost no decrease in activity and no increase in particle size were observed even after 10 h of the water-splitting reaction (Fig. 29D(b)). Moreover, it was confirmed that both the reverse reaction (Fig. 20C) and O2-photoreduction reaction (Fig. 20D) were well suppressed over this sample, as expected.270 These results indicate that loading Rh2−xCrxO3 NCs using our method is very effective for creating highly functional water-splitting photocatalysts.
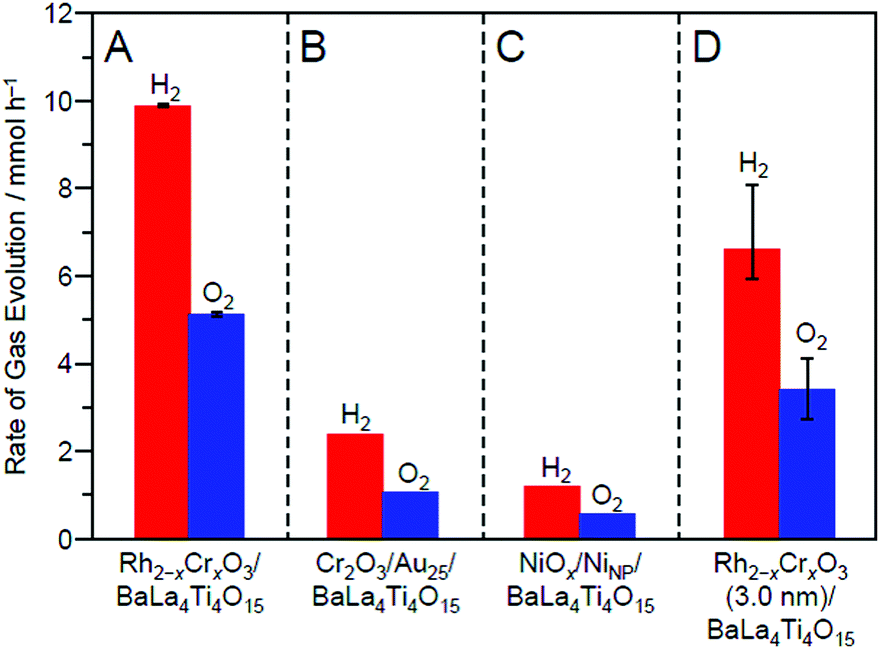 | ||
| Fig. 30 Comparison of the rates of H2 and O2 evolution by photocatalytic water-splitting over different photocatalysts: (A) Rh2−xCrxO3/BaLa4Ti4O15 (0.09 wt% Rh and 0.10 wt% Cr), (B) Cr2O3/Au25/BaLa4Ti4O15 (0.10 wt% Au and 0.50 wt% Cr), (C) NiOx/NiNP/BaLa4Ti4O15 (0.50 wt% Ni), and (D) Rh2−xCrxO3 (3.0 nm)/BaLa4Ti4O15 (0.10 wt% Rh and 0.15 wt% Cr). In this study, NiOx/NiNP/BaLa4Ti4O15 and Rh2−xCrxO3 (3.0 nm)/BaLa4Ti4O15 loaded with Rh2−xCrxO3 NPs of ∼3.0 nm were prepared by the impregnation method. Reproduced with permission from ref. 270. Copyright 2020 Wiley-VCH. | ||
The method of loading Rh2−xCrxO3 NCs established in this study can be applied to other photocatalysts in principle. In addition, Rh2−xCrxO3 is an effective cocatalyst for various water-splitting photocatalysts.266–269,271,272 In the future, it is expected that high quantum yields can be achieved for many water-splitting photocatalysts using this loading method.
4. Conclusions
We have attempted to create practical water-splitting photocatalysts by combining the most advanced techniques of both metal NCs and water-splitting photocatalysts (Fig. 4). The following findings were obtained regarding the loading of an atomically precise cocatalyst, the electronic/geometric structure of a cocatalyst, the correlation between the types of loaded metal NCs and the water-splitting activity, and the functionalization of a water-splitting photocatalyst.Loading of atomically precise metal NCs
(i) When Aun(SG)m NCs are used as precursors, it is possible to load Aun NCs with a controlled number of constituent atoms on BaLa4Ti4O15 and SrTiO3.(ii) To load Aun NCs while maintaining the number of constituent atoms of the precursor NCs, it is indispensable to use Aun(SG)m NCs with high stability in solution as the precursor.
(iii) If hydrophobic metal NCs are used as precursors, replacing some of the ligands of the NCs with hydrophilic ligands via ligand exchange is an effective means of loading atomically precise metal NCs on the photocatalyst.
(iv) An increase in the amount of loaded NCs induces an increase in the particle size of the loaded NCs. In the loading of Aun (n = 10, 15, 18, 25, or 39), Au24Pd, and Au24Pt on BaLa4Ti4O15, when metal NCs are loaded with <0.2 wt% metal, it is possible to suppress the aggregation of NCs on the photocatalysts.
Electronic structures
(i) The loaded metal NCs have electronic structures different from those of the precursor metal NCs.(ii) In Au25/BaLa4Ti4O15, partial charge transfer occurs from BaLa4Ti4O15 to Au25.
(iii) Substitution of Au in Au25/BaLa4Ti4O15 with Pd or Pt increases the electron density of Au in the loaded NCs.
Geometric structures
(i) On BaLa4Ti4O15, Au25 NCs have a relatively planar geometric structure, and Au24Pd and Au24Pt NCs have more three-dimensional structures.(ii) In Au24Pd/BaLa4Ti4O15, Pd is located on the surface of the loaded metal NCs and bound to S. On the other hand, in Au24Pt/BaLa4Ti4O15, Pt is located at the interface between Au24Pt NCs and BaLa4Ti4O15.
(iii) In Au25/BaLa4Ti4O15 and Au24Pd/BaLa4Ti4O15, there are only a few Au–O bonds at the interface between the metal NCs and the photocatalyst, leading to the weak immobilization of the loaded metal NCs on the photocatalyst. On the other hand, in Au24Pt/BaLa4Ti4O15, several Pt–O bonds are formed at the interface of the metal NC/photocatalyst, leading to stronger immobilization of the loaded metal NCs on the photocatalyst.
Catalytic activity
(i) With the same Au loading amount, Aun/BaLa4Ti4O15 with a smaller number of constituent atoms exhibits higher photocatalytic activity.(ii) The difference in activity among Aun/BaLa4Ti4O15 (n = 10, 15, 18, 25, and 39; 0.1 wt% Au) is mainly due to the change in the proportion of surface Au atoms.
(iii) The higher activity of Aun/BaLa4Ti4O15 compared with AuNP/BaLa4Ti4O15 is caused by the increase in the number of surface Au atoms that exceeds the decrease in activity per Au atom.
(iv) The size effect of the cocatalyst changes depending on the type of flowing gas. When using flowing gas not containing CO2, the water-splitting activity is not changed by mere miniaturization of the Au cocatalyst. This phenomenon is explained by the acceleration of both the HER and O2-reduction reaction due to miniaturization of the Au cocatalyst.
(v) The substitution of one Au of Au25/BaLa4Ti4O15 with Pd induces a decrease in the water-splitting activity. On the other hand, the substitution of one Au of Au25/BaLa4Ti4O15 with Pt induces an increase in the water-splitting activity. These opposite trends are caused by the difference in the substitution positions of Pd and Pt.
Functionalization
(i) A highly active water-splitting photocatalyst that takes advantage of the high H2-production ability of ultrafine Aun NCs can be created by forming a Cr2O3 shell on the surface of Aun NCs. When applying Pt substitution to such cocatalysts, further high water-splitting activity can be obtained.(ii) When using fine Rh2−xCrxO3 NCs as a cocatalyst, further enhancement of the activity can be achieved for BaLa4Ti4O15 photocatalysts.
It is expected that these findings will lead to clear design guidelines for the creation of practical water-splitting photocatalysts.
5. Outlook
The following efforts are considered necessary to obtain a deeper knowledge of water-splitting photocatalysts and thereby create practical photocatalysts.Establishment of a synthesis method of atomically precise Rhn NCs
The study on Aun/BaLa4Ti4O15, where the number of constituent atoms of the cocatalyst was controlled with atomic precision, revealed the details of the effect of miniaturization of the Au cocatalyst on each reaction. However, although the number of constituent atoms of the cocatalyst was not controlled with atomic precision, even higher water-splitting activity was achieved for Rh2−xCrxO3 NC/BaLa4Ti4O15. Therefore, precise synthesis methods are expected to be established for Rhn NCs in the future. If the obtained precise Rhn NCs would be used as precursors of cocatalysts, we could obtain deeper understanding of the functionalization of Rh2−xCrxO3 NC/BaLa4Ti4O15 and thereby create even more active Rh2−xCrxO3 NC/BaLa4Ti4O15.Use of non-noble metal cocatalysts
As shown in this feature article, the use of cocatalysts consisting of noble metals, such as Au, Pt, and Rh, is effective for producing high activity. However, these metals are expensive elements. Since previous studies showed that non-noble elements, such as nickel, cobalt, manganese, etc.,210,222,273 can also be used for the creation of active cocatalysts, research searching for effective non-noble metal cocatalysts is expected to be conducted in future studies. To achieve this, various precise synthesis methods are also expected to be established for non-noble metal NCs.274Selective loading of metal NCs on the optimal crystal plane
In a semiconductor photocatalyst, there are respective crystal planes which electrons and holes generated by photoexcitation can easily reach. If HER and OER cocatalysts could be selectively loaded on such crystal planes, electrons and holes could be efficiently used in the reaction.37,275 Thus, techniques for selectively loading metal NCs on specific crystal planes are expected to be developed in the future.Elucidation of the HER and OER activities of existing metal NCs using electrochemical methods
The research field of metal NCs has already realized the precise synthesis of many metal NCs. Among them, there might be metal NCs with electronic structures suitable for HER and OER cocatalysts. Because the HER and OER activity of each metal NC can be estimated by electrochemical measurements,21 it is expected that electrochemical experiments will be widely conducted for the existing precise metal NCs. These studies could lead to the discovery of novel high-performance cocatalysts.Elucidation of the geometric structures of loaded metal NCs
To understand the structure–property relationship, it is essential to obtain deeper understanding of the geometrical structure of the loaded metal NCs. Therefore, in the future, it is expected that the geometric structure of the loaded metal NCs will be studied using Cs-corrected TEM276 and scanning TEM. In addition, operando measurements using X-ray absorption fine structure (XAFS) analysis277 are also expected to be conducted to obtain a deeper understanding of the geometric structure during the water-splitting reaction.Structural control of loaded metal NCs
Even if SR-protected metal NCs having no variation in the geometric structure are used as the precursor, there appears to be variation in the geometric structures of the loaded metal NCs. To identify the geometric structures of NCs that create high activity and to selectively load such NCs on the photocatalysts, it is necessary to establish methods to control the geometric structure of the loaded NCs. Previous studies have established various methods to refine the size and structure of SR-protected metal NCs dispersed in solution.153,158,193,278 In the future, it is expected that methods to refine the size and structure will also be established for the loaded NCs.Decreasing the size of loaded metal NCs
As shown in Section 3.1.1, decreasing the size of cocatalysts leads to higher activity. Recent studies by another group implied that atomically dispersed metal cocatalysts are also effective for improving water-splitting photocatalysts.279–281 In the future, it is expected that cocatalysts will be controlled in a smaller size region by using smaller ligand-protected metal clusters114,282–285 and thereby the effective size of the cocatalysts will be elucidated in more detail.Elucidation of the charge-transfer rate at the interface between the photocatalysts and cocatalysts
To obtain a highly active water-splitting photocatalyst, it is also important to increase the rate of carrier transfer and the carrier injection efficiency from the photocatalyst to the cocatalyst.286 However, currently, there is little information on these properties for photocatalysts loaded with fine metal NCs. In the future, it is expected that deeper understanding of the carrier transfer rate and carrier injection efficiency will be obtained through fluorescence lifetime measurements and transient absorption spectroscopy.66–72 Such information would lead to clearer design guidelines for the creation of high-performance cocatalysts that can effectively transfer excited electrons from photocatalysts to cocatalysts.Activation of visible-light-responsive photocatalysts
BaLa4Ti4O15 and SrTiO3 used in our research exhibit high apparent quantum yields and stabilities among ultraviolet light-driven water-splitting photocatalysts (left half part in Fig. 7). However, approximately 40% of solar energy is in the visible-light region. Therefore, effective use of visible light is indispensable for the practical application of water-splitting photocatalysts.78 Currently, there are only a few semiconductor photocatalysts that can completely split water in one step using visible light, such as GaN:ZnO, g-C3N4, etc. (right half part in Fig. 7).78 However, overall water-splitting using visible light can also be achieved by a photocatalytic system that uses a two-step reaction called the Z-scheme, which imitates photosynthesis in plants.25,26,29,31–34 In the future, it is expected that both of these visible-light-responsive water-splitting photocatalysts will be more activated based on the knowledge obtained in previous studies.Guidance by DFT calculations
As described above, in recent years, it has become possible to control the chemical composition of cocatalysts with atomic precision. For photocatalysts loaded with such atomically precise cocatalysts, DFT calculations could predict highly functional photocatalytic systems.287 In the future, it is expected that research aimed at the improvement of the functionality of photocatalysts will shift from trial-and-error experiments to prediction by DFT calculations.We hope that many water-splitting photocatalysts for practical use will be created by overcoming these issues and that we can welcome a society in which energy and environmental problems have been solved as soon as possible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank all the co-authors listed in the references. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant number 20H02698 and 20H02552), Scientific Research on Innovative Areas “Coordination Asymmetry” (grant number 17H05385 and 19H04595), and Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion” (grant number 18H05178 and 20H05115). Funding from the Asahi Glass Foundation, TEPCO Memorial Foundation Research Grant (Basic Research), and Kato Foundation for Promotion of Science (grant number KJ-2904) is gratefully acknowledged.References
- R. S. Stein and J. Powers, The Energy Problem, World Scientific,Singpore, 2011 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-e. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS.
- D. Eguchi, M. Sakamoto and T. Teranishi, Chem. Sci., 2018, 9, 261–265 RSC.
- K. Kwak, W. Choi, Q. Tang, D.-e. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS.
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC.
- L. Wang, Z. Tang, W. Yan, H. Yang, Q. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 20635–20641 CrossRef CAS.
- L. Sumner, N. A. Sakthivel, H. Schrock, K. Artyushkova, A. Dass and S. Chakraborty, J. Phys. Chem. C, 2018, 122, 24809–24817 CrossRef CAS.
- T. C. Jones, L. Sumner, G. Ramakrishna, M. b. Hatshan, A. Abuhagr, S. Chakraborty and A. Dass, J. Phys. Chem. C, 2018, 122, 17726–17737 CrossRef CAS.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 CrossRef CAS.
- T. Kawawaki, Y. Negishi and H. Kawasaki, Nanoscale Adv., 2020, 2, 17–36 RSC.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, 1802234 CrossRef.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543–544 RSC.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83–86 CrossRef CAS.
- J. M. Lehn, J. P. Sauvage and R. Ziessel, Nouv. J. Chim., 1980, 623–627 CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4317–4318 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- Y. Zhu, T. Wang, T. Xu, Y. Li and C. Wang, Appl. Surf. Sci., 2019, 464, 36–42 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS.
- T. Pradeep, NANO: The Essentials: Understanding Nanoscience and Nanotechnology, McGraw-Hill Education, New York, 2007 Search PubMed.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319–3329 CrossRef CAS.
- W. D. Knight, W. A. de Heer and W. A. Saunders, Z. Phys. D: At., Mol. Clusters, 1986, 3, 109–114 CrossRef CAS.
- T. P. Martin, T. Bergmann, H. Göehlich and T. Lange, J. Phys. Chem., 1991, 95, 6421–6429 CrossRef CAS.
- L.-S. Wang, O. Cheshnovsky, R. E. Smalley, J. P. Carpenter and S. J. Hwu, J. Chem. Phys., 1992, 96, 4028–4031 CrossRef CAS.
- R. L. Whetten, D. M. Cox, D. J. Trevor and A. Kaldor, Phys. Rev. Lett., 1985, 54, 1494–1497 CrossRef CAS.
- S. C. Richtsmeier, E. K. Parks, K. Liu, L. G. Pobo and S. J. Riley, J. Chem. Phys., 1985, 82, 3659–3665 CrossRef CAS.
- P. Fayet, F. Granzer, G. Hegenbart, E. Moisar, B. Pischel and L. Wöste, Z. Phys. D: At., Mol. Clusters, 1986, 3, 299–302 CrossRef CAS.
- A. Nakajima, K. Hoshino, T. Naganuma, Y. Sone and K. Kaya, J. Chem. Phys., 1991, 95, 7061–7066 CrossRef CAS.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, R. Arai, Y. Niihori and T. Tsukuda, Chem. Commun., 2011, 47, 5693–5695 RSC.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- W. Kurashige and Y. Negishi, J. Cluster Sci., 2012, 23, 365–374 CrossRef CAS.
- Y. Negishi, U. Kamimura, M. Ide and M. Hirayama, Nanoscale, 2012, 4, 4263–4268 RSC.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- Y. Negishi, W. Kurashige, Y. Kobayashi, S. Yamazoe, N. Kojima, M. Seto and T. Tsukuda, J. Phys. Chem. Lett., 2013, 4, 3579–3583 CrossRef CAS.
- Y. Negishi, W. Kurashige, Y. Niihori and K. Nobusada, Phys. Chem. Chem. Phys., 2013, 15, 18736–18751 RSC.
- A. Puls, P. Jerabek, W. Kurashige, M. Förster, M. Molon, T. Bollermann, M. Winter, C. Gemel, Y. Negishi, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 4327–4331 CrossRef CAS.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- H. N. Ghosh, J. B. Asbury and T. Lian, J. Phys. Chem. B, 1998, 102, 6482–6486 CrossRef CAS.
- B. Ohtani, R. M. Bowman, D. P. Colombo Jr., H. Kominami, H. Noguchi and K. Uosaki, Chem. Lett., 1998, 579–580 CrossRef CAS.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Chem. Phys. Lett., 2001, 333, 271–277 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- J. Lee, H. S. Shim, M. Lee, J. K. Song and D. Lee, J. Phys. Chem. Lett., 2011, 2, 2840–2845 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- X.-Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2018, 3, 46–52 CrossRef CAS.
- M. R. Nellist, J. Qiu, F. A. L. Laskowski, F. M. Toma and S. W. Boettcher, ACS Energy Lett., 2018, 3, 2286–2291 CrossRef CAS.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS.
- T. Tsukuda and H. Häkkinen, Protected Metal Clusters: From Fundamentals to Applications. Elsevier, Amsterdam, 2015 Search PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS.
- T. Tsukuda, Bull. Chem. Soc. Jpn., 2012, 85, 151–168 CrossRef CAS.
- B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104–3113 CrossRef CAS.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem. – Eur. J., 2020, 26, 16150–16193 CrossRef CAS.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- S. Bhat, A. Baksi, S. K. Mudedla, G. Natarajan, V. Subramanian and T. Pradeep, J. Phys. Chem. Lett., 2017, 8, 2787–2793 CrossRef CAS.
- S. Tian, L. Liao, J. Yuan, C. Yao, J. Chen, J. Yang and Z. Wu, Chem. Commun., 2016, 52, 9873–9876 RSC.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284–25290 CrossRef CAS.
- C. Kumara, C. M. Aikens and A. Dass, J. Phys. Chem. Lett., 2014, 5, 461–466 CrossRef CAS.
- C. Yao, Y.-j. Lin, J. Yuan, L. Liao, M. Zhu, L.-h. Weng, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 15350–15353 CrossRef CAS.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Häkkinen, B. K. Teo and N. Zheng, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS.
- W. Kurashige, S. Yamazoe, M. Yamaguchi, K. Nishido, K. Nobusada, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 2072–2076 CrossRef CAS.
- S. Sharma, W. Kurashige, K. Nobusada and Y. Negishi, Nanoscale, 2015, 7, 10606–10612 RSC.
- Y. Niihori, M. Eguro, A. Kato, S. Sharma, B. Kumar, W. Kurashige, K. Nobusada and Y. Negishi, J. Phys. Chem. C, 2016, 120, 14301–14309 CrossRef CAS.
- Y. Niihori, S. Hossain, B. Kumar, L. V. Nair, W. Kurashige and Y. Negishi, APL Mater., 2017, 5, 053201 CrossRef.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D.-e. Jiang and Y. Negishi, Nanoscale, 2018, 10, 18969–18979 RSC.
- S. Hossain, Y. Imai, D. Suzuki, W. Choi, Z. Chen, T. Suzuki, M. Yoshioka, T. Kawawaki, D. Lee and Y. Negishi, Nanoscale, 2019, 11, 22089–22098 RSC.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horiz., 2020, 7, 796–803 RSC.
- S. Hossain, T. Ono, M. Yoshioka, G. Hu, M. Hosoi, Z. Chen, L. V. Nair, Y. Niihori, W. Kurashige, D.-e. Jiang and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 2590–2594 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS.
- W. Kurashige, K. Munakata, K. Nobusada and Y. Negishi, Chem. Commun., 2013, 49, 5447–5449 RSC.
- W. Kurashige, S. Yamazoe, K. Kanehira, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2013, 4, 3181–3185 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 4134–4142 CrossRef CAS.
- S. Hossain, W. Kurashige, S. Wakayama, B. Kumar, L. V. Nair, Y. Niihori and Y. Negishi, J. Phys. Chem. C, 2016, 120, 25861–25869 CrossRef CAS.
- Y. Niihori, S. Hossain, S. Sharma, B. Kumar, W. Kurashige and Y. Negishi, Chem. Rec., 2017, 17, 473–484 CrossRef CAS.
- X. Kang and M. Zhu, Small, 2019, 15, 1902703 CrossRef CAS.
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS.
- J.-J. Li, Z.-J. Guan, Z. Lei, F. Hu and Q.-M. Wang, Angew. Chem., Int. Ed., 2019, 58, 1083–1087 CrossRef CAS.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334 RSC.
- E. G. Mednikov and L. F. Dahl, Philos. Trans. R. Soc., A, 2010, 368, 1301–1332 CrossRef CAS.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- M. Schulz-Dobrick and M. Jansen, Z. Anorg. Allg. Chem., 2007, 633, 2326–2331 CrossRef CAS.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS.
- J. D. Roth, G. J. Lewis, L. K. Safford, X. Jiang, L. F. Dahl and M. J. Weaver, J. Am. Chem. Soc., 1992, 114, 6159–6169 CrossRef CAS.
- A. Ceriotti, N. Masciocchi, P. Macchi and G. Longoni, Angew. Chem., Int. Ed., 1999, 38, 3724–3727 CrossRef CAS.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, J. Cluster Sci., 2014, 25, 115–146 CrossRef CAS.
- S. S. Kurasov, N. K. Eremenko, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1989, 361, 405–408 CrossRef CAS.
- L. Hao, G. J. Spivak, J. Xiao, J. J. Vittal and R. J. Puddephatt, J. Am. Chem. Soc., 1995, 117, 7011–7012 CrossRef CAS.
- E. Cattabriga, I. Ciabatti, C. Femoni, T. Funaioli, M. C. Iapalucci and S. Zacchini, Inorg. Chem., 2016, 55, 6068–6079 CrossRef CAS.
- C. Cesari, I. Ciabatti, C. Femoni, M. C. Iapalucci, F. Mancini and S. Zacchini, Inorg. Chem., 2017, 56, 1655–1668 CrossRef CAS.
- L. V. Nair, S. Hossain, S. Wakayama, S. Takagi, M. Yoshioka, J. Maekawa, A. Harasawa, B. Kumar, Y. Niihori, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2017, 121, 11002–11009 CrossRef CAS.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem. DOI:10.1021/acs.inorgchem.0c00879.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS.
- B. K. Teo and H. Zhang, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 5067–5071 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106–109 RSC.
- Y. Negishi, Bull. Chem. Soc. Jpn., 2014, 87, 375–389 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- V. G. Albano, P. L. Bellon, M. Manassero and M. Sansoni, J. Chem. Soc. D, 1970, 1210–1211 RSC.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. H. M. Calis and J. W. A. van der Velden, Chem. Ber., 1981, 114, 3634–3642 CrossRef CAS.
- P. Chini, J. Organomet. Chem., 1980, 200, 37–61 CrossRef CAS.
- M. Paolieri, I. Ciabatti and M. Fontani, J. Cluster Sci., 2019, 30, 1623–1631 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- A. Ebina, S. Hossain, H. Horihata, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS.
- Y. Niihori, K. Yoshida, S. Hossain, W. Kurashige and Y. Negishi, Bull. Chem. Soc. Jpn., 2019, 92, 664–695 CrossRef CAS.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- H. Yu, B. Rao, W. Jiang, S. Yang and M. Zhu, Coord. Chem. Rev., 2019, 378, 595–617 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- T. G. Schaaff, G. Knight, M. N. Shafigullin, R. F. Borkman and R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646 CrossRef CAS.
- S. Kumar, M. D. Bolan and T. P. Bigioni, J. Am. Chem. Soc., 2010, 132, 13141–13143 CrossRef CAS.
- K. Kimura, N. Sugimoto, S. Sato, H. Yao, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 14076–14082 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS.
- C. Gautier and T. Bürgi, J. Am. Chem. Soc., 2006, 128, 11079–11087 CrossRef CAS.
- Y. Negishi, S. Hashimoto, A. Ebina, K. Hamada, S. Hossain and T. Kawawaki, Nanoscale, 2020, 12, 8017–8039 RSC.
- V. L. Jimenez, M. C. Leopold, C. Mazzitelli, J. W. Jorgenson and R. W. Murray, Anal. Chem., 2003, 75, 199–206 CrossRef CAS.
- S. Knoppe and P. Vogt, Anal. Chem., 2019, 91, 1603–1609 CrossRef CAS.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS.
- Y. Negishi, C. Sakamoto, T. Ohyama and T. Tsukuda, J. Phys. Chem. Lett., 2012, 3, 1624–1628 CrossRef CAS.
- D. M. Black, S. B. H. Bach and R. L. Whetten, Anal. Chem., 2016, 88, 5631–5636 CrossRef CAS.
- D. M. Black, N. Bhattarai, S. B. H. Bach and R. L. Whetten, J. Phys. Chem. Lett., 2016, 7, 3199–3205 CrossRef CAS.
- Y. Niihori, M. Matsuzaki, T. Pradeep and Y. Negishi, J. Am. Chem. Soc., 2013, 135, 4946–4949 CrossRef CAS.
- Y. Niihori, M. Matsuzaki, C. Uchida and Y. Negishi, Nanoscale, 2014, 6, 7889–7896 RSC.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS.
- Y. Niihori, C. Uchida, W. Kurashige and Y. Negishi, Phys. Chem. Chem. Phys., 2016, 18, 4251–4265 RSC.
- Y. Niihori, Y. Kikuchi, D. Shima, C. Uchida, S. Sharma, S. Hossain, W. Kurashige and Y. Negishi, Ind. Eng. Chem. Res., 2017, 56, 1029–1035 CrossRef CAS.
- Y. Niihori, D. Shima, K. Yoshida, K. Hamada, L. V. Nair, S. Hossain, W. Kurashige and Y. Negishi, Nanoscale, 2018, 10, 1641–1649 RSC.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS.
- Y. Niihori, S. Hashimoto, Y. Koyama, S. Hossain, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2019, 123, 13324–13329 CrossRef CAS.
- A. Ghosh, J. Hassinen, P. Pulkkinen, H. Tenhu, R. H. A. Ras and T. Pradeep, Anal. Chem., 2014, 86, 12185–12190 CrossRef CAS.
- C. Yao, J. Chen, M.-B. Li, L. Liu, J. Yang and Z. Wu, Nano Lett., 2015, 15, 1281–1287 CrossRef CAS.
- S. Hossain, D. Suzuki, T. Iwasa, R. Kaneko, T. Suzuki, S. Miyajima, Y. Iwamatsu, S. Pollitt, T. Kawawaki, N. Barrabés, G. Rupprechter and Y. Negishi, J. Phys. Chem. C, 2020, 124, 22304–22313 CrossRef CAS.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 1999, 103, 9394–9396 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS.
- A. C. Dharmaratne, T. Krick and A. Dass, J. Am. Chem. Soc., 2009, 131, 13604–13605 CrossRef CAS.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D.-e. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS.
- S. Takano, S. Yamazoe, K. Koyasu and T. Tsukuda, J. Am. Chem. Soc., 2015, 137, 7027–7030 CrossRef CAS.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS.
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS.
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, J. Am. Chem. Soc., 2015, 137, 9511–9514 CrossRef CAS.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS.
- R. Kazan, U. Müller and T. Bürgi, Nanoscale, 2019, 11, 2938–2945 RSC.
- Y. Li, M. Chen, S. Wang and M. Zhu, Nanomaterials, 2018, 8, 1070 CrossRef.
- R. Kazan, B. Zhang and T. Bürgi, Dalton Trans., 2017, 46, 7708–7713 RSC.
- S. Yang, J. Chai, Y. Song, J. Fan, T. Chen, S. Wang, H. Yu, X. Li and M. Zhu, J. Am. Chem. Soc., 2017, 139, 5668–5671 CrossRef CAS.
- R. L. Whetten, M. N. Shafigullin, J. T. Khoury, T. G. Schaaff, I. Vezmar, M. M. Alvarez and A. Wilkinson, Acc. Chem. Res., 1999, 32, 397–406 CrossRef CAS.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef.
- R. L. Whetten and R. C. Price, Science, 2007, 318, 407–408 CrossRef CAS.
- Y. Song, S. Wang, J. Zhang, X. Kang, S. Chen, P. Li, H. Sheng and M. Zhu, J. Am. Chem. Soc., 2014, 136, 2963–2965 CrossRef CAS.
- J. Zhong, X. Tang, J. Tang, J. Su and Y. Pei, J. Phys. Chem. C, 2015, 119, 9205–9214 CrossRef CAS.
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, J. Am. Chem. Soc., 2015, 137, 652–655 CrossRef CAS.
- S. Ito, S. Takano and T. Tsukuda, J. Phys. Chem. Lett., 2019, 10, 6892–6896 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Energy Environ. Sci., 2009, 2, 306–314 RSC.
- K. Maeda and K. Domen, Chem. Mater., 2010, 22, 612–623 CrossRef CAS.
- T. Tomio, H. Miki, H. Tabata, T. Kawai and S. Kawai, J. Appl. Phys., 1994, 76, 5886–5890 CrossRef CAS.
- T. Puangpetch, S. Chavadej and T. Sreethawong, Energy Convers. Manage., 2011, 52, 2256–2261 CrossRef CAS.
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000–1030 CrossRef CAS.
- Y. Miseki, S. Fujiyoshi, T. Gunji and K. Sayama, J. Phys. Chem. C, 2017, 121, 9691–9697 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem. – Eur. J., 2010, 16, 7750–7759 CrossRef CAS.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467–2473 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS.
- A. Xiong, T. Yoshinaga, T. Ikeda, M. Takashima, T. Hisatomi, K. Maeda, T. Setoyama, T. Teranishi and K. Domen, Eur. J. Inorg. Chem., 2014, 767–772 CrossRef CAS.
- T. Yoshinaga, M. Saruyama, A. Xiong, Y. Ham, Y. Kuang, R. Niishiro, S. Akiyama, M. Sakamoto, T. Hisatomi, K. Domen and T. Teranishi, Nanoscale, 2018, 10, 10420–10427 RSC.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS.
- K. Nakata, S. Sugawara, W. Kurashige, Y. Negishi, M. Nagata, S. Uchida, C. Terashima, T. Kondo, M. Yuasa and A. Fujishima, Int. J. Photoenergy, 2013, 2013, 456583 CrossRef.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- N. W. Pirie and K. G. Pinhey, J. Biol. Chem., 1929, 84, 321–333 CAS.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. C, 2008, 112, 19797–19800 CrossRef CAS.
- D.-e. Jiang, M. Kühn, Q. Tang and F. Weigend, J. Phys. Chem. Lett., 2014, 5, 3286–3289 CrossRef CAS.
- W. Kurashige, R. Kumazawa, Y. Mori and Y. Negishi, J. Mater. Appl., 2018, 7, 1–11 Search PubMed.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- M. P. Johansson, A. Lechtken, D. Schooss, M. M. Kappes and F. Furche, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 77, 053202 CrossRef.
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116, 4094–4101 CrossRef CAS.
- A. Lechtken, C. Neiss, M. M. Kappes and D. Schooss, Phys. Chem. Chem. Phys., 2009, 11, 4344–4350 RSC.
- W. Huang and L.-S. Wang, Phys. Rev. Lett., 2009, 102, 153401 CrossRef.
- H. Häkkinen, B. Yoon, U. Landman, X. Li, H.-J. Zhai and L.-S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef.
- H. Häkkinen and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R2287–R2290 CrossRef.
- D.-e. Jiang, S. H. Overbury and S. Dai, J. Phys. Chem. Lett., 2011, 2, 1211–1215 CrossRef CAS.
- G. Zanti and D. Peeters, J. Phys. Chem. A, 2010, 114, 10345–10356 CrossRef CAS.
- H. K. Yuan, A. L. Kuang, C. L. Tian and H. Chen, AIP Adv., 2014, 4, 037107 CrossRef.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, Florida, 2007 Search PubMed.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- A. A. Melvin, K. Illath, T. Das, T. Raja, S. Bhattacharyya and C. S. Gopinath, Nanoscale, 2015, 7, 13477–13488 RSC.
- S.-F. Hung, Y.-C. Yu, N.-T. Suen, G.-Q. Tzeng, C.-W. Tung, Y.-Y. Hsu, C.-S. Hsu, C.-K. Chang, T.-S. Chan, H.-S. Sheu, J.-F. Lee and H. M. Chen, Chem. Commun., 2016, 52, 1567–1570 RSC.
- H. Bian, N. T. Nguyen, J. Yoo, S. Hejazi, S. Mohajernia, J. Müller, E. Spiecker, H. Tsuchiya, O. Tomanec, B. E. Sanabria-Arenas, R. Zboril, Y. Y. Li and P. Schmuki, ACS Appl. Mater. Interfaces, 2018, 10, 18220–18226 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Chem. – Eur. J., 2013, 19, 4986–4991 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- E. J. Braunschweig, A. D. Logan, A. K. Datye and D. J. Smith, J. Catal., 1989, 118, 227–237 CrossRef CAS.
- A. D. Logan, E. J. Braunschweig, A. K. Datye and D. J. Smith, Langmuir, 1988, 4, 827–830 CrossRef CAS.
- S. Labich, E. Taglauer and H. Knözinger, Top. Catal., 2001, 14, 153–161 CrossRef.
- L. Liu, C. Ge, W. Zou, X. Gu, F. Gao and L. Dong, Phys. Chem. Chem. Phys., 2015, 17, 5133–5140 RSC.
- X. Liu, M.-H. Liu, Y.-C. Luo, C.-Y. Mou, S. D. Lin, H. Cheng, J.-M. Chen, J.-F. Lee and T.-S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef CAS.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS.
- T. Ohno, L. Bai, T. Hisatomi, K. Maeda and K. Domen, J. Am. Chem. Soc., 2012, 134, 8254–8259 CrossRef CAS.
- K. Maeda, D. Lu, K. Teramura and K. Domen, Energy Environ. Sci., 2010, 3, 471–478 RSC.
- W. Kurashige, Y. Mori, S. Ozaki, M. Kawachi, S. Hossain, T. Kawawaki, C. J. Shearer, A. Iwase, G. F. Metha, S. Yamazoe, A. Kudo and Y. Negishi, Angew. Chem., Int. Ed., 2020, 59, 7076–7082 CrossRef CAS.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- T. H. Chiang, H. Lyu, T. Hisatomi, Y. Goto, T. Takata, M. Katayama, T. Minegishi and K. Domen, ACS Catal., 2018, 8, 2782–2788 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS.
- J. Ji, G. Wang, T. Wang, X. You and X. Xu, Nanoscale, 2014, 6, 9185–9191 RSC.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef.
- R. Takahata, S. Yamazoe, Y. Maehara, K. Yamazaki, S. Takano, W. Kurashige, Y. Negishi, K. Gohara and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 6907–6912 CrossRef CAS.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176–184 CrossRef CAS.
- S. Tian, Y. Cao, T. Chen, S. Zang and J. Xie, Chem. Commun., 2020, 56, 1163–1174 RSC.
- Y. H. Li, J. Xing, X. H. Yang and H. G. Yang, Chem. – Eur. J., 2014, 20, 12377–12380 CrossRef CAS.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem. – Eur. J., 2014, 20, 2138–2144 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS.
- J. Chen, L. Liu, X. Liu, L. Liao, S. Zhuang, S. Zhou, J. Yang and Z. Wu, Chem. – Eur. J., 2017, 23, 18187–18192 CrossRef CAS.
- S. R. Biltek, S. Mandal, A. Sen, A. C. Reber, A. F. Pedicini and S. N. Khanna, J. Am. Chem. Soc., 2013, 135, 26–29 CrossRef CAS.
- S. R. Biltek, A. Sen, A. F. Pedicini, A. C. Reber and S. N. Khanna, J. Phys. Chem. A, 2014, 118, 8314–8319 CrossRef CAS.
- S. R. Biltek, A. C. Reber, S. N. Khanna and A. Sen, J. Phys. Chem. A, 2017, 121, 5324–5331 CrossRef CAS.
- X. L. Du, X. L. Wang, Y. H. Li, Y. L. Wang, J. J. Zhao, L. J. Fang, L. R. Zheng, H. Tong and H. G. Yang, Chem. Commun., 2017, 53, 9402–9405 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |