
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric [N–I–N]+ halonium complexes in solution?†
Daniel
von der Heiden
 a,
Kari
Rissanen
b and
Máté
Erdélyi
*a
a,
Kari
Rissanen
b and
Máté
Erdélyi
*a
aDepartment of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. E-mail: mate.erdelyi@kemi.uu.se
bUniversity of Jyvaskyla, Department of Chemistry, 40014 Jyväskylä, Finland
First published on 27th October 2020
Abstract
Assessment of the solution equilibria of [bis(pyridine)iodine(I)]+ complexes by ESI-MS and NMR reveals the preference of iodine(I) to form complexes with a more basic pyridine. Mixtures of symmetric [bis(pyridine)iodine(I)]+ complexes undergo statistical ligand exchange, with a predominant entropic driving force favoring asymmetric systems. The influence of ligand basicity, concentration, temperature, and ligand composition is evaluated. Our findings are expected to facilitate the investigations, and the supramolecular and synthetic applications of halonium ions’ halogen bonds.
A halogen bond is the net attractive interaction of a polarized halogen and a Lewis base.1 Even though this phenomenon has been studied since the 1860s,2 it has first recently received increased interest and gained applications, for instance in supramolecular chemistry,3,4 materials sciences,5–7 drug design,8–10 and organic synthesis.11–14 [Bis(pyridine)iodine]BF4 ((1)2I, Fig. 1), became a common synthetic reagent (Barluenga reagent).11,12 This complex is held together by a three-center, four-electron [N–I–N]+ halogen bond, in which the halogen bond donor iodine(I) is stabilized by two Lewis basic nitrogen atoms simultaneously.15 The binding free energy of such an interaction is typically ΔG ≥ −100 kJ mol−1,15,16 higher than that of conventional halogen bonding systems possessing a neutral halogen bond donor (ΔG < −24 kJ mol−1).17–20
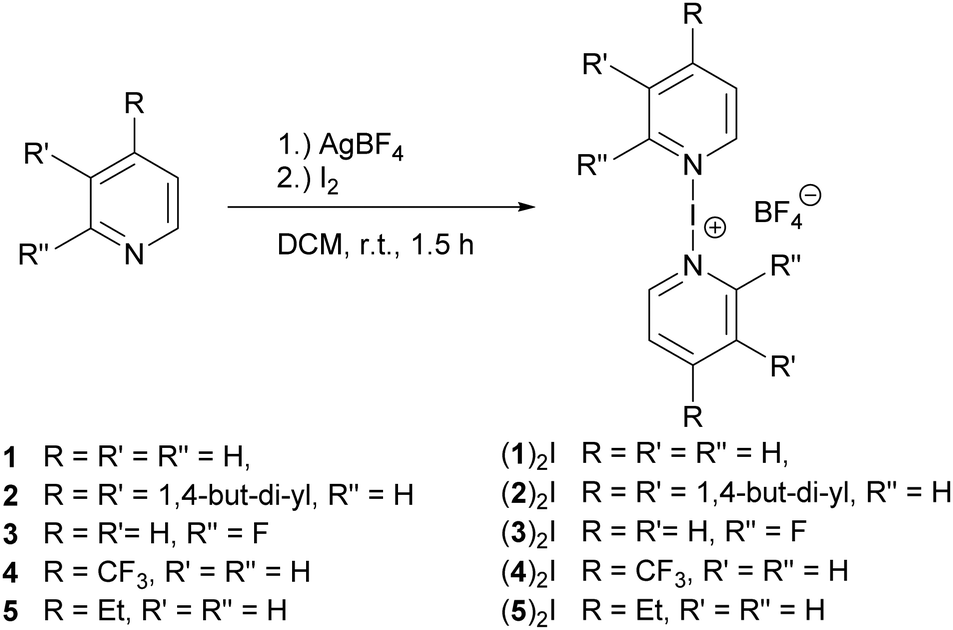 | ||
| Fig. 1 Synthesis of the [bis(pyridine)iodine(I)]+ complexes (1–5)2I. | ||
The [N–I–N]+ halogen bond shows a preference for forming symmetric systems.15,16,21 The first asymmetric [bis(pyridine)-iodine(I)]+-type complex has been achieved by iodine(I) complexation to a bidentate ligand that was designed to offer halogen bond acceptors with distinctly different electron densities, whereas preventing ligand scrambling by the use of a rigid backbone connecting the two pyridines.23 A recent report provided the first solid-state proof for unrestrained asymmetric [bis(pyridine)iodine(I)]+-type complexes.22 These were synthesized from a mixture of different pyridines, presuming a preference for selective formation of the asymmetric complex, and its trapping into solid-state by crystallization (Fig. 2).
 | ||
| Fig. 2 Reported synthesis of an asymmetric [bis(pyridine)iodine(I)]BF4 complex.22 | ||
Motivated by the above study, we report herein the assessment of ligand-exchange in [N–I–N]+ halogen bond complexes, and of the preference of formation of symmetric versus asymmetric halogen bond arrangements in solution.
Synthesis. The iodine(I) complexes (1–5)2I (Fig. 1) were prepared by mixing 2.0 eq. of the corresponding pyridine with 1.0 equivalent of AgBF4 in CH2Cl2. Upon adding 1.05 equivalent of Iodine to this solution, a precipitate formed. A purple colour, caused by the presence of a slight excess of free iodine, indicated the completion of the reaction. The solution was filtered, and a colourless [N–I–N]+ complex obtained by precipitation upon addition of a 3-fold volume of n-hexane.
[Bis(pyridine)iodine(I)]+-type complexes in the presence of free ligand. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [bis(5,6,7,8-tetrahydroiso-quinoline)2iodine(I)]BF4 complex ((2)2I) and free 5,6,7,8-tetrahydroiso-quinoline (2) gave well-resolved 1H NMR signals (Fig. 3a–c), indicating the presence of two species in solution. To evaluate whether a potential ligand exchange24 takes place, a 1D-NOESY experiment using a selective excitation-pulse at 7.24 ppm, H−5 of (2)2I, was performed. Observation of 13% magnetisation transfer from the ligand of the (2)2I complex to the H−5 resonance of the free ligand 2 revealed ligand exchange, in CD2Cl2 solution at room temperature.
:1 mixture of [bis(5,6,7,8-tetrahydroiso-quinoline)2iodine(I)]BF4 complex ((2)2I) and free 5,6,7,8-tetrahydroiso-quinoline (2) gave well-resolved 1H NMR signals (Fig. 3a–c), indicating the presence of two species in solution. To evaluate whether a potential ligand exchange24 takes place, a 1D-NOESY experiment using a selective excitation-pulse at 7.24 ppm, H−5 of (2)2I, was performed. Observation of 13% magnetisation transfer from the ligand of the (2)2I complex to the H−5 resonance of the free ligand 2 revealed ligand exchange, in CD2Cl2 solution at room temperature.
 | ||
| Fig. 3
1H (500 MHz, CD2Cl2, 298 K) of (a) [bis(5,6,7,8-tetrahydroisoquinoline)iodine(I)]BF4 ((2)2I); (b) of 5,6,7,8-tetrahydroisoquinoline (2); (c) 1:2 mixture of (2)2I and 2; (d) selective 1H NOESY NMR of the mixture with a selective pulse at 7.24 ppm. | ||
Next, [bis(2-fluoropyridine)iodine(I)]BF4 ((3)2I) and 2-fluoropyridine (3) as well as their 1:1 mixture was studied (Fig. 4). Pyridine 3 was selected for its exceptional weak Lewis basicity (pKa(3H+) = −0.44),25 expectably resulting in formation of a weak [N–I–N]+ halogen bond complex. The 1H and 13C NMR spectra of (3)2I showed broad signals, and the addition of 3 lead to further linebroadening (Fig. S8 and S18, ESI†). In contrast to the mixture of 2 and (2)2I, we could not observe separate sets of signals for the free ligand (3) and the corresponding [N–I–N]+ complex (3)2I. The signal coalescence revealed fast ligand exchange of 3. The different behaviour of the two halogen bond complexes is explained by a higher exchange rate of the (3)2I complex, formed by the weaker halogen bond acceptor 3, as compared to that of the (2)2I complex of the stronger acceptor 2. In addition to the exchange rate, the point of signal coalescence is further influenced by the chemical shift separation of the signals of the free ligand and of that in its iodine(I) complex, and by spectrometer frequency, as shown in Sections S2.b–d of the ESI.†
 | ||
| Fig. 4
1H (500 MHz, CD3CN, 298 K) of (a) pyridine (1); (b) (1)2I; (c) a 1.0:1.1 mixture of (3)2I and (1); (d) (3)2I; (e) and free 2-fluoropyridine (3). Fast exchanging ligand 3 (green, dots). | ||
[Bis(pyridine)iodine(I)]+-type complexes in the presence of a different pyridine ligand. Upon addition of 1.1 eq. pyridine (1) to [bis(2-fluoropyridine)iodine(I)]BF4 ((3)2I), the appearance of a new species (Fig. 4c, red) was observed along with the disappearance of the signals corresponding to free pyridine (1). Traces of (1)2I could be observed (Fig. 4c, blue), in addition to broad signals resembling those seen for the rapidly exchanging 2-fluoropyridine (3) within its free, (3)2I and (1)1(3)1I forms (Fig. 4, green dots; for (3)/(3)2I exchange, see Fig. S18, ESI†). The appearance of the new signals, marked with red in Fig. 4c, indicates the formation of an asymmetric (1)1(3)1I complex in solution, proving ligand exchange in the halogen bond complex. The deshielding of the signals of (1)1(3)1I are consistent with the chemical shift changes that were previously reported upon formation of an analogous asymmetric [bis(pyridine)iodine]+-type complex.23
Next, we added various amounts of pyridine (1) to a CD2Cl2 solution of [bis(4-trifluoromethylpyridine)iodine(I)]BF4 ((4)2I) (Section S3.b, ESI†). Upon addition of >2 eq. of 1, signals consistent with (1)2I, 4 and 1 were observed, whereas signals corresponding to (4)2I could not be detected. This indicates that upon addition of the stronger halogen bond acceptor pyridine (1), the weaker halogen bond acceptor 4-trifluoromethylpyridine (4) was replaced in the iodine(I) complex. When less than <2 eq. of pyridine (1) was added, a mixture of (1)2I, (4)2I and (1)1(4)1I along with 4-trifluoromethylpyridine (4) and pyridine (1) were observed by comparison of their 1H and 15N NMR chemical shifts to that of the pure compounds. The mixed (1)1(4)1I complex was identified by comparison of its 15N NMR chemical shifts (15N = −156 ppm and −184 ppm) to those of (1)2I (15N = −174 ppm) and (4)2I (15N = −167 ppm). The shielding and deshielding of the 15N NMR signals of (1)1(4)1I as compared to (1)2I and (4)2I are comparable to those observed for a similar asymmetric [bis(pyridine)iodine(I)]+-type complex (Fig. 5).23 We also observed mass signals for (1)2I, (1)1(4)1I, and (4)2I as well as for their fragments (1)1I and (4)1I in a 1:1 mixture of (1)2I and (4)2I in solution by ESI-MS (Fig. S30 and S31, ESI†).
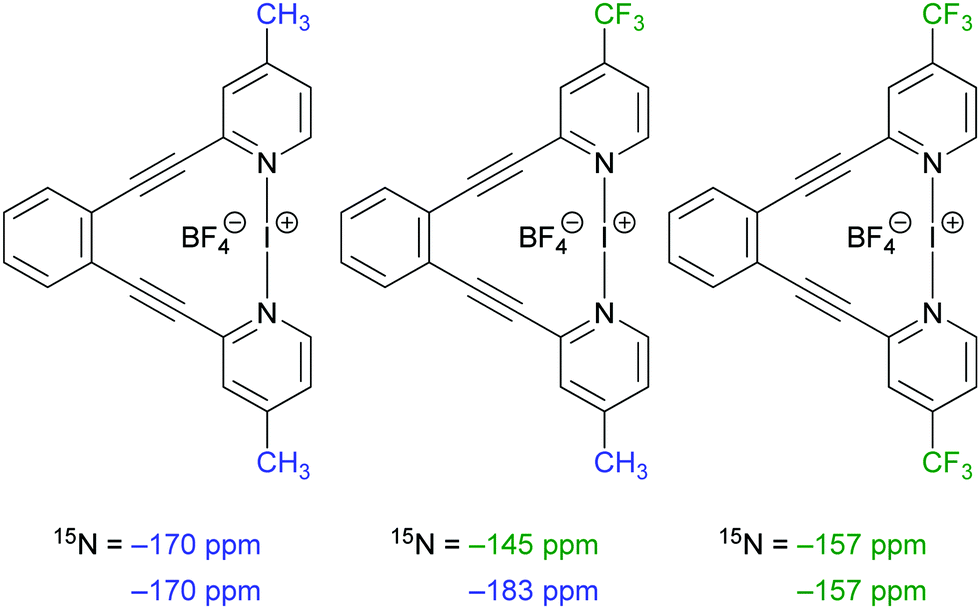 | ||
| Fig. 5 15N NMR shifts of selected symmetric and asymmetric [bis(pyridine)iodine(I)]+-type complexes formerly published by our group illustrate the effect of asymmetric substitution on the resulting pyridine 15N NMR resonance frequency.23 | ||
The molar ratios of (1)2I, (1)1(4)1I and (4)2I in CD2Cl2 solutions prepared by mixing (1)2I and (4)2I in different ratios (0.1:0.9, 0.3:0.7, 0.5:0.5, 0.7:0.3, 0.9:0.1) was evaluated by integration of the corresponding 1H NMR signals (Fig. 6). The observed statistical distribution of the species indicates a dominant entropic contribution at 25 °C in the formation of the asymmetric (1)1(4)1I complex from (1)2I and (4)2I. The equilibrium observed at 25 °C was unaffected by temperature change (5 °C–35 °C), twenty-fold dilution, or addition of 0.88 eq. of the less basic 4-trifluoromethylpyridine (4) (Sections 3.c–h, ESI†). Having this in hand we tested our hypothesis on a 1:1 mixture of (1)2I and (5)2I to compare our results with the previously reported findings (Fig. 2). Indeed we could observe the presence of a statistical 1:2:1 mixture of (1)2I:(1)1(5)1I:(5)2I using 1H NMR, 13C, and 15N NMR (Section 3.i, ESI†). The entropy-driven ligand exchange in mixtures of symmetric [bis(pyridine)iodine(I)]+-type complexes takes place and should also be anticipated for asymmetric [bis(pyridine)iodine(I)]+-type complexes in solution, due to the principle of microscopic reversibility.
 | ||
| Fig. 6 Observed mixture (y-axis) after mixing different molar fractions of (4)2I in (1)2I (x-axis). On the y-achses, the resulting molar fractions of (1)2I (red), (1)1(4)1I (green), (4)2I (blue) as well as ratios expected in case of an statistical distribution of ligand 1 and 4 (black lines) are depicted. | ||
This work proves that the halogen bond complexes of iodonium ions undergo chemical exchange, with the exchange rate dependent on the Lewis basicity of the halogen bond acceptor. Their NMR spectra depend on experimental factors, such as the spectrometer frequency, chemical shift separation as well as the lineshape of the exchanging peaks (ESI†). The exchange is revealed by line broadening for the complexes of weak halogen bond acceptors, whereas it is more difficult to observe for those of stronger acceptors. For the latter, the observation of a single set of separate signals for the two halogen bond acceptors involved does not necessarily indicate the presence of a single, mixed complex in solution, but is rather the result of the slow exchange of statistically distributed complexes. Even if the mixed species can be crystallized from such a solution,22 this may not indicate that only the mixed complex would be present in solution. Accordingly, bidentate model systems, which prohibit the scrambling of Lewis bases,6,23 such as those shown in Fig. 5, have great advantages over pyridine complexes for fundamental studies, such as IPE NMR.16,23
We conclude that a higher pyridine basicity correlates with a lower ligand exchange rate, and that the mixing of two symmetric halogen bonded iodine(I) complexes leads to statistical distribution of the pyridine ligands across the iodenium ions. These findings are expected to support future investigations of the halogen bond complexes of halonium ions as well as their synthetic applications.
This project made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy, and was supported by Vinnova [2019-02160], FORMAS [2017-0009] and the Swedish Research Council [2016-03602].
Conflicts of interest
There are no conflicts to declare.References
- G. Desiraju, P. Ho and L. Kloo, et al. , Pure Appl. Chem., 2013, 85, 1711 CAS.
- F. Guthrie, J. Chem. Soc., 1863, 16, 239 RSC.
- K. Rissanen, CrystEngComm, 2008, 10, 1107 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281 CrossRef CAS.
- D. W. Bruce, P. Metrangolo, F. Meyer, T. Pilati, C. Präsang, G. Resnati, G. Terraneo, S. G. Wainwright and A. C. Whitwood, Chem. – Eur. J., 2010, 16, 9511 CrossRef CAS.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030 CrossRef CAS.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A.-C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870 CrossRef CAS.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267 RSC.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez and J. Benz, Angew. Chem., Int. Ed., 2011, 50, 314 CrossRef CAS.
- J. Barluenga, J. M. González, P. J. Campos and G. Asensio, Angew. Chem., Int. Ed. Engl., 1985, 24, 319 CrossRef.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Chem. – Eur. J., 2004, 10, 4206 CrossRef CAS.
- D. von der Heiden, E. Detmar, R. Kuchta and M. Breugst, Synlett, 2018, 1307 CAS.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622 CrossRef CAS.
- L. Turunen and M. Erdelyi, Chem. Soc. Rev., 2020, 49, 2688 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706 CrossRef CAS.
- M. Breugst, D. von der Heiden and J. Schmauck, Synthesis, 2017, 3224 CAS.
- C. Laurence, J. Graton, M. Berthelot and M. J. El Ghomari, Chem. – Eur. J., 2011, 17, 10431 CrossRef CAS.
- M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646 CrossRef CAS.
- O. Dumele, D. Wu, N. Trapp, N. Goroff and F. Diederich, Org. Lett., 2014, 16, 4722 CrossRef CAS.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdelyi, Chem. Commun., 2012, 48, 1458 RSC.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428 RSC.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 13503 CrossRef CAS.
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, Aust. J. Chem., 2013, 66, 1179 CrossRef CAS.
- S. Zhang, J. Comput. Chem., 2012, 33, 2469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and details of synthesis. See DOI: 10.1039/d0cc06706g |
| This journal is © The Royal Society of Chemistry 2020 |