
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A photocaged inhibitor of acid sphingomyelinase†
Kevin
Prause
a,
Gita
Naseri
 a,
Fabian
Schumacher
bc,
Christian
Kappe
a,
Burkhard
Kleuser
b and
Christoph
Arenz
*a
a,
Fabian
Schumacher
bc,
Christian
Kappe
a,
Burkhard
Kleuser
b and
Christoph
Arenz
*a
aInstitute for Chemistry, Humboldt Universität zu Berlin, 12437 Berlin, Germany. E-mail: arenzchr@hu-berlin.de
bInstitute of Pharmacy, Freie Universität Berlin, Königin-Luise-Str. 2+4, 14195 Berlin, Germany
cDepartment of Molecular Biology, University of Duisburg-Essen, Hufelandstr. 55, 45147 Essen, Germany
First published on 6th November 2020
Abstract
Acid sphingomyelinase (ASM) is a potential drug target and involved in rapid lipid signalling events. However, there are no tools available to adequately study such processes. Based on a non cell-permeable PtdIns(3,5)P2 inhibitor of ASM, we developed a compound with o-nitrobenzyl photocages and butyryl esters to transiently mask hydroxyl groups. This resulted in a potent light-inducible photocaged ASM inhibitor (PCAI). The first example of a time-resolved inhibition of ASM was shown in intact living cells.
Sphingolipids are ubiquitous and important in eukaryotic cells as structural and signaling lipids.1,2 Due to the lipid nature of the intermediates, investigation into this class of metabolites is not straight forward, and usually requires tedious extraction procedures. As a consequence, topological as well as time-resolution is often lost during their analysis, although the exact localization of sphingolipids is key to their function. This fundamental problem has made research on sphingolipids largely dependent on functionalized lipid analogues, including radioactively or fluorescently labeled compounds,3 cross-link compounds,4,5 or photo-switchable versions of metabolites.6 Acid sphingomyelinase (ASM) is an important enzyme involved in sphingolipid metabolism, as it cleaves the major plasma membrane lipid sphingomyelin during lysosomal membrane digestion.7 In addition to this constitutive degradation process, the activity of ASM can be stimulated by various exogenic triggers such as cytokines, ionizing radiation and reactive oxygen species (ROS). As a result, recent evidence suggests a fusion of lysosomes with the plasma membrane within seconds after stimulation being the major source of a secreted form of ASM8,9 Rapid cleavage of sphingomyelin results in transient and local elevation of downstream metabolites, such as ceramide and sphingosine, which are likely responsible for ASM-mediated effects. ASM is being discussed as a drug target for treating acute-lung injury,10 sepsis,11 metastasis of melanoma12 and major depression.13 However, no potent drug-like inhibitors of ASM are known.14 Currently, functional inhibitors of acid sphingomyelinase (FIASMA) are the first choice to inhibit ASM, although they are known to be indirect and non-specific.15 The bisphosphonate Arc39 is the most potent inhibitor known.16 The compound acts on ASM with high potency in cells, but not in mice.16 Recently, a novel hydroxamic acid inhibitor showed antidepressant activity in a mouse model,17 but not much is known about its specificity.
One of the first ASM inhibitors discovered, is the naturally occurring phosphatidyl inositol-3,5-bisphosphate (PtdIns(3,5)P2), but its structure prevents its use in cell culture or in vivo.18 Our goal was to synthesize a cell-compatible analogue of PtdIns(3,5)P2, based on the previously published analogue rac-12.19 In the latter compound, the phosphatidic acid is replaced by a simple sulfonic ester (Fig. 1). We now wanted to mask the phosphate groups at the 3′ and 5′ position with non-polar residues to allow efficient penetration of cells.
 | ||
| Fig. 1 Stepwise modification of the initial natural compound inhibitor PtdIns3,5-bP, into 1-O-sulfonyl-myo-inositol rac-12,19 the photocaged phosphoramide 1 and the completely masked analogue 2 (PCAI). | ||
We were well aware of the fact that such a masking strategy would also offer the possibility of installing photo-labile groups, enabling a light-induced inhibition.
Such photocaging strategies are ideal to study lipid-signaling events, as they provide the spatiotemporal resolution needed.
Indeed, photocaged biologically active compounds like photo-caged enzyme inhibitors have shown to be invaluable tools to study biological phenomena.20 For sphingolipid biology, a number of light-activated probes have been developed, including caged21 and photo-switchable6 sphingosine-1-phosphate, photocaged sphingosine,22 photocaged ceramide23 and ceramide-1-phosphate.24 However, to the best of our knowledge no tools for light-activated control over ASM activity exist.
For the synthesis of the envisioned photocaged inhibitor, we planned to replace the phosphoester bonds found in PtdIns(3,5)P2 and our previously synthesized analogues rac-12 (Fig. 1) by chemically less stable phosphoramidate bonds. Half-life times of phosphoramidates under mild acidic pH values are typically in the range of 30 minutes up to a few hours.25 Due to the acidic pH in the lysosomes, this could potentially cause a transient inhibition rather than a long-term depletion of ASM, like it is observed for FIASMAs. This would potentially be more in line with the idea of a time-resolved inhibition.
We therefore planned the synthesis of compound 1 carrying photolabile masking groups of the phosphoramidate moieties to facilitate cell penetration (Fig. 1). As compound 1 – like other phosphoinositides – would be still a rather polar molecule, we planned in addition, to synthesize compound 2, which is also masked at the hydroxyl groups by biologically labile butyl ester groups, similar to earlier described cell-permeable phosphoinositides.26
For synthesis of the desired compounds, the literature-known 3,5-diamino-D-myo-inositol 10 was synthesized from N-acetyl-D-glucosamine (GlcNAc) in ten steps, following a route described by Ogawa et al. (Scheme S1, ESI†).27 Compound 10 was converted into the novel bisazide 11 by copper(II) – mediated diazo transfer. Careful sulfonylation yielded the desired regioisomer 12a as a main product (Scheme 1).
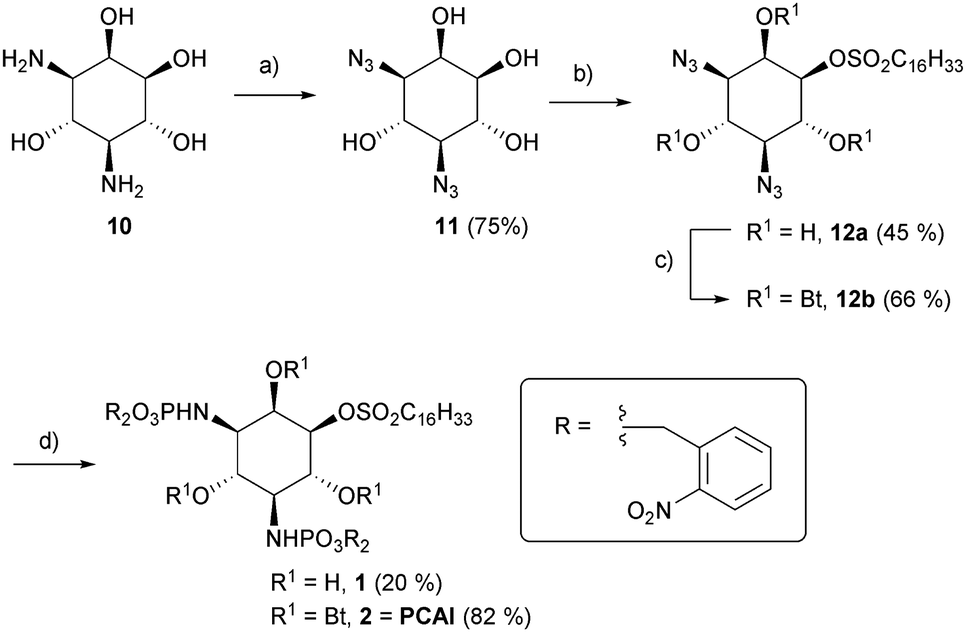 | ||
Scheme 1 (a) N3SO2Im*HCl, Cu(II)SO4, K2CO3, rt, 12 h; (b) ClSO2C16H33, DMAP, THF, 0 °C to RT, 2 h (c) pyridine, Bt2O, 0 °C to rt, 2 d. (d) (1) P(ONB)3, CH2Cl2, 45–50 °C, 24–50 h (2) MeCN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) | ||
The latter was either directly subjected to a Staudinger phosphite reaction28 to afford 1 or previously treated with butyric acid anhydride to obtain the more lipophilic compound 2. The overall synthetic yields starting from 10 (Scheme S1, ESI†) were 6.7% (1) and 18.3% (2), respectively.
After having accomplished the synthesis of the desired compounds, inhibition of ASM by the UV-treated compounds was tested. As expected, ASM inhibition was confirmed for uncaged phosphoramidite derived from 1 at 5 μM. Noteworthy, the IC50 value for ASM inhibition for the previously reported phosphoinositide mimic rac-12 was found to be 0.5 μM (C12 sulfonic ester)19 and ∼200 nM for the C16 sulfonic ester (unpublished results). In the next step, we wanted to monitor ASM inhibition in a continuous, time-resolved assay in vitro, making use of ASM FRET substrates, which have been published recently.29,30 It was clear to us that irradiation for the purpose of uncaging would also be associated with bleaching of the FRET probes. When compound 1 (10 μM) was incubated with ASM in presence of the visual range FRET probe,30 similar slopes for caged inhibitor-containing reaction and the control reaction were observed (Fig. 2). Then, both reactions were illuminated with UV light for one minute, respectively. As expected, bot reactions showed a drop in fluorescence as expected due to FRET probe bleaching, but now a clear difference in slopes, indicating ASM activity, was observed.
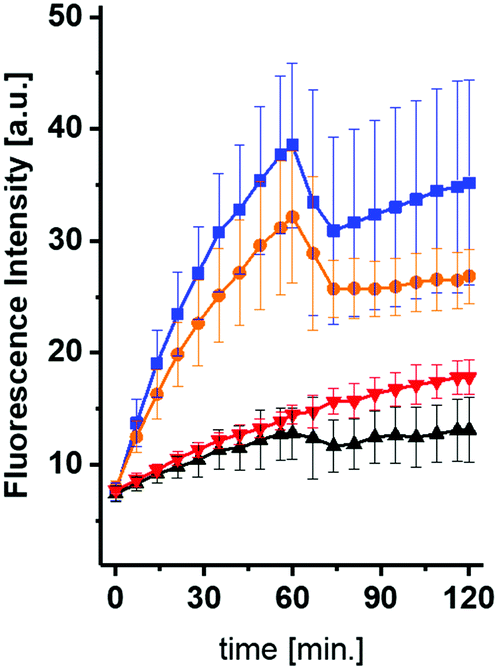 | ||
| Fig. 2 In vitro de-caging experiment in real time using 10 μM of compound 1. After 60 minutes, the reactions were illuminated for 60 seconds. Orange: 1; blue: w/o inhibitor. Red: reaction with 1, but illuminated before. Black = red, but with an additional illumination. | ||
While the fluorescence intensity for the reaction without inhibitor increased further, the fluorescence for reaction containing compound 1 remained almost constant. The slope for the latter reaction was now similar to the slope for the reaction containing 1, which was illuminated beforehand.
However, it was also clearly visible, that even the caged compound 1 showed some inhibition at this concentration. These observations suggested that a minor portion of fully or partially uncaged inhibitor was formed either during handling or during excitation of the FRET probe.
In order to investigate this more thoroughly, we performed experiments using 10 μM (Fig. S7, ESI†) or 25 μM (Fig. S8, ESI†) of compound 1, which was illuminated prior to ASM assay for different durations. Indeed, even illumination times as short as 5 seconds led to significantly increased inhibitions. However, 25 μM of 1 not illuminated prior to reaction led to about 50% inhibition, suggestion that (based on IC50 of rac-12) roughly 5% of 1 were uncaged during handling or excitation of the FRET probe (Fig. S8, ESI†). We also studied the uncaging kinetics and the stability of 2 under the assay conditions in more detail and found that half-life times for uncaging in vitro were ∼10 s (5 μM) and ∼100 s (5 mM). The isolated yield for 2 was 93% (Fig. S1, ESI†). No decomposition of the phosphoamidate bonds at pH 4.5 or pH 7.4 was observed even after 48 h (Fig. S2–S5, ESI†). When incubated for 30 minutes in cell lysates, a mixture of 2 and 1 but no other degradation products were recovered by HPLC analysis (Fig. S6, ESI†).
Next, we wanted to test the photocaged compounds in living cells. For analysis of ASM activity, we made use of the visible range FRET probe in combination with flow cytometry, as published recently.30 To this end, we incubated HEK293 cells with different concentrations of both photocaged inhibitors. In order to avoid photo-bleaching of the ASM probe, we decided to incubate the cells with the caged inhibitors for a 16 h, then induce uncaging of the inhibitors by a light pulse and finally add the FRET probe for another 16 h to the cells for an assessment of ASM activity. First, inhibition of both probes was tested at concentrations of 25 μM and 50 μM, respectively (Fig. 3).
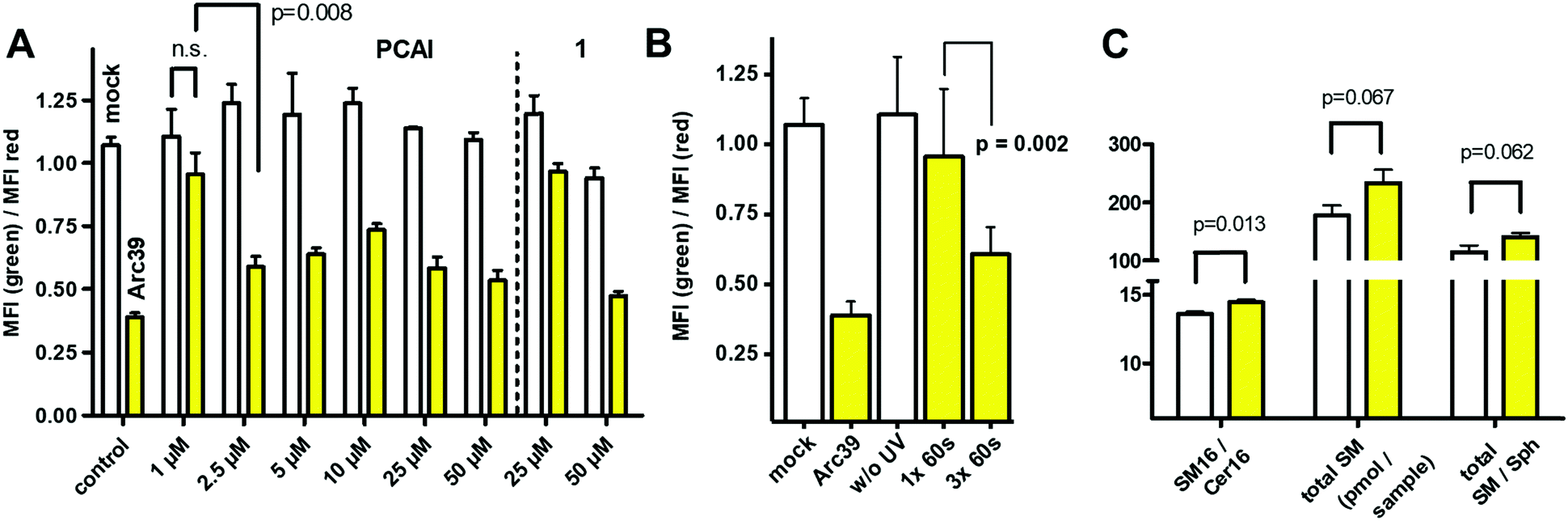 | ||
| Fig. 3 (A) Photo-induced inhibition of ASM in living cells. Monitored by flow cytometry as the ratio of geometric mean fluorescence intensities (MFI) in green and red channels. White columns: without photo-induction, yellow columns: with photo-induction (30 s). Control: white column: mock treated, yellow column: 10 μM Arc39, leading almost complete ASM inhibition. If not indicated, all differences between respective white and yellow columns are highly significant (p < 0.001). n.s. = not significant. (B) Inhibition of ASM in living cells, using PCAI (1 μM) without or with illumination for 1× 60 s or 3× 60 s. (C) Changes to the HEK293 lipidome in presence of PCAI (10 μM, 60 s illumination, followed by 24 h chase). All experiments were performed in three distinct cell cultures as three technical replicates. SM = sphingomyelin, Cer16 = ceramide (N-palmitoylsphingosine), Sph = sphingosine. | ||
For probe 1, only a slight but significant inhibition was observed at 25 μM after irradiation, while inhibition at 50 μM was close to the inhibition shown by Arc39 (10 μM), which again was assumed to be close to 100%, based on previous experiments.16 Notably, without illumination, ASM activity in presence of 1 at 25 μM appeared to be normal, but with a slight but significant reduction in activity at 50 μM. This could reflect the behavior of 1in vitro, which also showed some inhibition at 25 μM, even in the presence of the photocage. In comparison to 1, compound 2 appeared to be more potent and therefore lower concentrations were investigated (Fig. 3A). Indeed, between 2.5 μM and 50 μM, almost full inhibition of ASM was observed after 30 seconds of illumination, but with normal ASM activities when this step was omitted. Therefore we termed compound 2 as “photocaged ASM inhibitor” (PCAI). At 1 μM, only a moderate decrease in ASM activity was observed after irradiation, which was not significant. However, when PCAI was illuminated three times for 60 seconds (each), a highly significant inhibition was observed (Fig. 3B). In another experiment, we illuminated cells incubated with 5 μM of PCAI for 15, 30, 45, 60 and 90 seconds, respectively. This experiment not only showed a highly significant inhibition after only 15 seconds of illumination, but also that 60 seconds illumination was not enough to exploit the full inhibitory potential of PCAI. PCAI differs from 1 in butyryl groups masking the hydroxyl groups of the inositol ring. We assume, that superior potency of PCAI over 1 is due to a higher level of cell penetration, which in turn is due to higher polarity of intact 1, but may be also a result of partial decaging of 1.
In order to substantiate our finding that PCAI is a cell-permeable photo-activatable inhibitor of ASM, the effect of PCAI on cellular lipid levels was investigated, although a recent study showed that the overall effects are rather small, even in presence of 50 μM FIASMA (imipramine).31 Towards this end, we incubated HEK293 cells with 10 μM of the compound, followed by illumination for 60 seconds. Compared to cells devoid of the inhibitor, total sphingomyelin (SM) was increased (p = 0.06), as well as the ratios of total SM/Cer (p = 0.35) and total SM/Sph (p = 0.06), but none of these values increased significantly. In contrast, SM16/Cer16 ratio was increased significantly (p = 0.01), which represent the main substrate/product pair of ASM (Fig. 3C). These results are remarkable, given the fact that SM is one of the most abundant plasma membrane lipids and does not change its total concentration rapidly.
In conclusion, we have succeeded in developing the first photo-caged ASM inhibitor (PCAI). While decaged compound 1 shows full activity only at high concentrations with significant background activity, PCAI appeared to be much more potent, obviously due to better cell penetration. It is well possible that compound 1 is partially uncaged during experiments with the FRET probe employed for ASM monitoring. The presence of the biologically labile butyryl groups was key to make PCAI suitable for real world applications. The compound shows highly significant inhibition of ASM in living cells after illumination. Although a single illumination for 60 s is not enough to achieve 100% uncaging, it is definitely enough for a pronounced and highly significant inhibition at most concentrations. Indeed, illumination time may be even shortened at higher inhibitor concentrations. In future, the PCAI approach might be further improved by introduction of a longer sulfonic ester “tail” or by photocages that combine photolability with lysosomal enrichment.22 Certainly, PCAI will expand the toolbox for sphingolipid research and should be suitable for exhibiting spatio-temporal control over ASM during lipid signaling in cell and tissue culture.
C. A. acknowledges financial support by the Deutsche Forschungsgemeinschaft (AR 376-12/2).
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Kolter and K. Sandhoff, Angew. Chem., Int. Ed., 1999, 38, 1532–1568 CrossRef CAS.
- Y. A. Hannun and L. M. Obeid, Nat. Rev. Mol. Cell Biol., 2018, 19, 175–191 CrossRef CAS.
- G. Schwarzmann, C. Arenz and K. Sandhoff, Biochim. Biophys. Acta, 2014, 1841, 1161–1173 CrossRef CAS.
- P. Haberkant and J. C. Holthuis, Biochim. Biophys. Acta, 2014, 1841, 1022–1030 CrossRef CAS.
- P. Haberkant, R. Raijmakers, M. Wildwater, T. Sachsenheimer, B. Brugger, K. Maeda, M. Houweling, A. C. Gavin, C. Schultz, G. van Meer, A. J. Heck and J. C. Holthuis, Angew. Chem., Int. Ed., 2013, 52, 4033–4038 CrossRef CAS.
- J. Morstein, R. Z. Hill, A. J. E. Novak, S. Feng, D. D. Norman, P. C. Donthamsetti, J. A. Frank, T. Harayama, B. M. Williams, A. L. Parrill, G. J. Tigyi, H. Riezman, E. Y. Isacoff, D. M. Bautista and D. Trauner, Nat. Chem. Biol., 2019, 15, 623–631 CrossRef CAS.
- T. Kolter and K. Sandhoff, Annu. Rev. Cell Dev. Biol., 2005, 21, 81–103 CrossRef CAS.
- N. W. Andrews, Cell. Microbiol., 2019, 21, e13065 CrossRef CAS.
- C. S. Ferranti, J. Cheng, C. Thompson, J. Zhang, J. A. Rotolo, S. Buddaseth, Z. Fuks and R. N. Kolesnick, J. Cell Biol., 2020, 219, e201903176 CrossRef CAS.
- R. Goggel, S. Winoto-Morbach, G. Vielhaber, Y. Imai, K. Lindner, L. Brade, H. Brade, S. Ehlers, A. S. Slutsky, S. Schutze, E. Gulbins and S. Uhlig, Nat. Med., 2004, 10, 155–160 CrossRef.
- H. Y. Chung, D. C. Hupe, G. P. Otto, M. Sprenger, A. C. Bunck, M. J. Dorer, C. L. Bockmeyer, H. P. Deigner, M. H. Graler and R. A. Claus, Mol. Med., 2016, 22, 412–423 CAS.
- A. Carpinteiro, K. A. Becker, L. Japtok, G. Hessler, S. Keitsch, M. Pozgajova, K. W. Schmid, C. Adams, S. Muller, B. Kleuser, M. J. Edwards, H. Grassme, I. Helfrich and E. Gulbins, EMBO Mol. Med., 2015, 7, 714–734 CrossRef CAS.
- E. Gulbins, M. Palmada, M. Reichel, A. Luth, C. Bohmer, D. Amato, C. P. Muller, C. H. Tischbirek, T. W. Groemer, G. Tabatabai, K. A. Becker, P. Tripal, S. Staedtler, T. F. Ackermann, J. van Brederode, C. Alzheimer, M. Weller, U. E. Lang, B. Kleuser, H. Grassme and J. Kornhuber, Nat. Med., 2013, 19, 934–938 CrossRef CAS.
- D. Canals, D. M. Perry, R. W. Jenkins and Y. A. Hannun, Br. J. Pharmacol., 2011, 163, 694–712 CrossRef CAS.
- J. Kornhuber, P. Tripal, M. Reichel, L. Terfloth, S. Bleich, J. Wiltfang and E. Gulbins, J. Med. Chem., 2008, 51, 219–237 CrossRef CAS.
- E. Naser, S. Kadow, F. Schumacher, Z. H. Mohamed, C. Kappe, G. Hessler, B. Pollmeier, B. Kleuser, C. Arenz, K. A. Becker, E. Gulbins and A. Carpinteiro, J. Lipid Res., 2020, 61, 896–910 CrossRef CAS.
- K. Yang, J. Yu, K. Nong, Y. Wang, A. Niu, W. Chen, J. Dong and J. Wang, J. Med. Chem., 2020, 63, 961–974 CrossRef CAS.
- M. Kolzer, C. Arenz, K. Ferlinz, N. Werth, H. Schulze, R. Klingenstein and K. Sandhoff, Biol. Chem., 2003, 384, 1293–1298 Search PubMed.
- A. G. Roth, S. Redmer and C. Arenz, ChemBioChem, 2009, 10, 2367–2374 CrossRef CAS.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS.
- L. Qiao, A. P. Kozikowski, A. Olivera and S. Spiegel, Bioorg. Med. Chem. Lett., 1998, 8, 711–714 CrossRef CAS.
- S. Feng, T. Harayama, D. Chang, J. T. Hannich, N. Winssinger and H. Riezman, Chem. Sci., 2019, 10, 2253–2258 RSC.
- Y. A. Kim, D. M. Ramirez, W. J. Costain, L. J. Johnston and R. Bittman, Chem. Commun., 2011, 47, 9236–9238 RSC.
- R. S. Lankalapalli, A. Ouro, L. Arana, A. Gomez-Munoz and R. Bittman, J. Org. Chem., 2009, 74, 8844–8847 CrossRef CAS.
- C. J. Choy, C. R. Ley, A. L. Davis, B. S. Backer, J. J. Geruntho, B. H. Clowers and C. E. Berkman, Bioconjugate Chem., 2016, 27, 2206–2213 CrossRef CAS.
- C. Schultz, Bioorg. Med. Chem., 2003, 11, 885–898 CrossRef CAS.
- S. Ogawa, K. L. Rinehart, G. Kimura and R. P. Johnson, J. Org. Chem., 1974, 39, 812–821 CrossRef CAS.
- R. Serwa, I. Wilkening, G. Del Signore, M. Muhlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234–8239 CrossRef CAS.
- T. Pinkert, D. Furkert, T. Korte, A. Herrmann and C. Arenz, Angew. Chem., Int. Ed., 2017, 56, 2790–2794 CrossRef CAS.
- C. Kappe, Z. H. Mohamed, E. Naser, A. Carpinteiro and C. Arenz, Chemistry, 2020, 26, 5780–5783 CrossRef CAS.
- M. J. Justice, I. Bronova, K. S. Schweitzer, C. Poirier, J. S. Blum, E. V. Berdyshev and I. Petrache, J. Lipid Res., 2018, 59, 596–606 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06661c |
| This journal is © The Royal Society of Chemistry 2020 |