
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusually selective synthesis of chlorohydrooligosilanes†
Thomas
Lainer
a,
Roland
Fischer
 a,
Mario
Leypold
a,
Michael
Holthausen
b,
Odo
Wunnicke
b,
Michael
Haas
*a and
Harald
Stueger
*a
a,
Mario
Leypold
a,
Michael
Holthausen
b,
Odo
Wunnicke
b,
Michael
Haas
*a and
Harald
Stueger
*a
aInstitute of Inorganic Chemistry, Technische Universität Graz, Stremayrgasse 9/IV, Graz 8010, Austria. E-mail: michael.haas@tugraz.at; harald.stueger@tugraz.at
bEvonik Creavis GmbH, Paul-Baumann-Strasse 1, Marl 45772, Germany
First published on 12th October 2020
Abstract
New pathways towards molecular chlorohydrooligosilanes enable their one-pot synthesis in preparative amounts either by the selective chlorination of the corresponding perhydrosilanes with HCl/AlCl3 or by the partial hydrogenation of perchlorooligosilanes with substoichiometric amounts of iBu2AlH. The unexpected selective formation of Cl3Si-substituted species in the partial hydrogenation reactions could be related to mechanistic aspects.
Chlorosilanes, especially chlorohydrosilanes, are ideal precursors for the generation of hyperpure polycrystalline silicon. Because of the dramatic rise of photovoltaic applications, the production capacity of polysilicon reached 600 kton per year at the end of 2018.1 The vast majority of polycrystalline Si produced worldwide is made by the chemical vapor deposition of HSiCl3.2 Less than 10% of all solar grade silicon is currently manufactured from silicon hydrides such as SiH4 in spite of their lower decomposition temperatures and the resulting lower energy cost incurred during the deposition process. The major drawbacks of silicon hydrides are their high production costs and difficulty in handling because of their pyrophoric nature. Therefore, the search for alternative precursor materials for silicon deposition is still an issue of primary importance.3
Based on calculations, two mechanisms for the deposition of silicon from HSiCl3 are possible. The disilane mechanism proposed by Swihart and Carr et al.4 and the radical mechanism by Cavallotti et al.5 In both mechanisms, the formation of disilanes as reaction intermediates plays a crucial role by enhancing the gas phase reactivity. This is due to their increased reactivity, determined by the relatively low Si–Si bond energy and small barriers for Cl and H intramolecular transfers. Therefore, it can be anticipated that chlorohydrooligosilanes will outperform the state-of-the-art precursors concerning their film deposition properties. A recently published study actually showed the formation of higher quality films at higher deposition rates using atomic layer deposition (ALD) processes for the precursor molecule HSi2Cl5 as compared to Si2Cl6.6
However, the state-of-the-art synthetic protocols of chlorohydrooligosilanes are not straightforward. Partial halogenation reactions of di-, tri- and tetrasilane with BX3, HX/AlX3, AgX, SnX4, HgX2 or X2 (X = Cl, Br, I) afforded product mixtures which could only be analyzed after further derivatization of the initial products.7 The partial hydrogenation of perchlorooligosilanes by treatment with substoichiometric amounts of LiAlH4 or Bu3SnH was also found to be rather unselective.8,9 Additionally, selected halohydrooligosilanes were prepared by the halodephenylation of appropriately substituted phenylated precursors with anhydrous HX.10 The challenge of this method is the preparation of the starting materials which frequently involves tedious multi-step procedures. In a more recent study, we observed the clean formation of 1,2,3,4-tetrachloroneopentasilane 2 after treatment of neopentasilane 1 with 3.5 equivalents of SnCl4.11 To the best of our knowledge this was the first example of a direct and selective functionalization of a higher silicon hydride on a preparative scale.
A major disadvantage of SnCl4 as a halogenating reagent is the formation of huge amounts of SnCl2, which are difficult to remove completely, particularly on a larger scale. Thus, we investigated the chlorination of neopentasilane with gaseous HCl in the presence of AlCl3 as the catalyst. When dry HCl gas was bubbled through a benzene solution of neopentasilane containing catalytic amounts of AlCl3 at 70 °C for 15 minutes, compound 2 was obtained with remarkable selectivity along with 1,2,3-trichloroneopentasilane 3 as the by-product (Scheme 1). Removal of the solvent and the catalyst and recondensation of the crude product afforded a mixture of 2 (85%) and 3 (15%) in >60% yield. Attempts to separate both components using distillation or crystallization were unsuccessful.
 | ||
| Scheme 1 Partial chlorination of neopentasilane with SnCl4 and HCl/AlCl3. | ||
An even more appealing approach to chlorohydrooligosilanes is the partial hydrogenation of perchlorooligosilanes with substoichiometric amounts of iBu2AlH. This pathway enables the synthesis of chlorohydrooligosilanes on a broader scope. In recent studies it has been found that iBu2AlH is suitable for the hydrogenation of linear and branched chlorooligosilanes under mild reaction conditions without the formation of any Si–Si bond scission products.12 When we reacted hexachlorodisilane (Si2Cl6), octachlorotrisilane (Si3Cl8), nonachloroisotetrasilane ClSi(SiCl3) or dodecachloroneopentasilane Si(SiCl3)4 with sub-stoichiometric quantities of iBu2AlH, we observed the predominant formation of the corresponding 1,1,1-trichlorooligosilanes 4–7 with unexpected selectivity (Table 1). SiHCl2- and SiH2Cl-moieties were only detected in minor side products. Based on screening experiments, we determined that the ideal amount of iBu2AlH equals the number of chlorine atoms minus three. If more equivalents of iBu2AlH were used, the completely hydrogenated species were formed as the major products. Application of less equivalents of iBu2AlH afforded increasing portions of higher chlorinated polysilanes.
| Precursor | Equiv. iBu2AlH | Molar ratio of main productsa | Yield of Cl3SiSinHma (%) |
|---|---|---|---|
| a Calculated by integrating 1H-NMR signals of the volatile fraction after trap-to-trap distillation. b Trap-to-trap distillation at room temperature and 0.01 mbar. c Trap-to-trap distillation at 50 °C and 0.01 mbar. d Fraction 1: trap-to-trap distillation at 50 °C and 0.01 mbar. e Fraction 2: subsequent trap-to-trap distillation of the high boiling fraction at 80 °C and 0.01 mbar. | |||
| Si2Cl6 | 3 | Cl3SiSiH3 (4) (54%); ClH2SiSiCl3 (40%); Si2H6 (6%)b | 39 |
| Si3Cl8 | 5 | Cl3SiSiH2SiH3 (5) (67%); Si3H8 (33%)b | 36 |
| ClSi(SiCl3)3 | 6 | Cl3SiSiH(SiH3)2 (6) (57%); HSi(SiH3)3 (43%)c | 28 |
| Si(SiCl3)4 | 9 | Cl3SiSi(SiH3)3 (7) (11%); Si(SiH3)4 (89%)d | 2 |
| Cl3SiSi(SiH3)3 (7) (67%); Si(SiH3)4 (33%)e | 28 | ||
This new synthetic method can be performed in the absence of any solvent at room temperature, which enabled the isolation of 4–7 along with the corresponding perhydrogenated species and small amounts of other Cl/H silanes using trap-to-trap distillation under vacuum. The structures and relative quantities of the individual species present in the volatile fraction were assigned using NMR spectroscopy. (The corresponding experimental data and procedures are included in the ESI.†) If Si2Cl6 was used as the precursor the volatile fraction additionally contained considerable amounts of 1,1,1,2-tetrachlorodisilane. In all other cases, higher chlorinated species could not be separated from iBu2AlCl and ended up in the high boiling residue of the vacuum condensation along with some unreacted starting material. For Si3Cl8 the addition of excess iBu2AlH to the higher boiling product fraction afforded a further 0.6 g of Si3H8 which means a more or less quantitative conversion of the starting material to chlorohydrotrisilanes. Extensive side reactions such as Si–Si bond scission or oligomerization, thus, can be ruled out. Yields calculated for 4–7 on the basis of the relative intensities of 1H-NMR signals range between 28 and 39%.
Very recently, neopentasilane and its derivatives have attracted considerable attention as precursors for the deposition of silicon and silicon heterostructures.13 Thus, we decided to study compound 7 in more detail. 7 could be isolated from the crude product mixture obtained according to Scheme 2 from the reaction of Si(SiCl3)4 with 9 equiv. of iBu2AlH by fractional distillation as a colorless oil in about 20% yield and characterized by comparing its NMR data with literature values.14 The resulting samples were reasonably pure but still contained small amounts of neopentasilane 1 and iBu2AlCl. For the isolation of pure 7 the application of more sophisticated distillation techniques would be necessary, which was beyond the scope of this study.
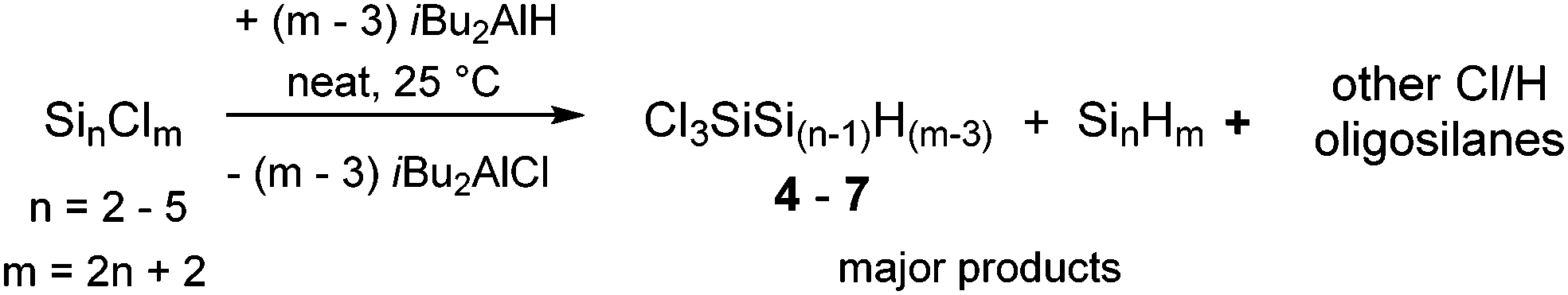 | ||
| Scheme 2 General scheme for the partial hydrogenation of perchlorooligosilanes with iBu2AlH. | ||
For nucleophilic substitution reactions of chlorosilanes a reactivity order of R3SiCl < R2SiCl2 < RSiCl3 < SiCl4 is usually found due to the impact of the electronegative Cl substituents at the silicon center.15 Attempts to partially hydrogenate polychlorosilanes with substoichiometric amounts of LiAlH4, thus, only afforded more or less statistical mixtures of various chlorohydrosilanes.8
The unexpected selectivity of the hydrogenation reactions with iBu2AlH towards the formation of 1,1,1-trichlorosilanes 4–7 as described above can be easily rationalized if mechanistic aspects are taken into account. Based on stereochemical arguments it has been proposed that the hydrogenation of chlorosilanes with iBu2AlH in non-coordinating solvents like hexane follows a SNi–Si-mechanism involving a four-center transition state A. In diethyl ether, on the contrary, the same hydrogenation reaction proceeds via a pentacoordinated transition state B typical for SN2 reactions (Scheme 3).16
 | ||
| Scheme 3 Mechanism of partial hydrogenation of 7. | ||
DFT calculations at the SMD (pentane) M06-2X-D3/aug-cc-pVTZ level of theory were performed on the reaction of Cl3SiSiCl3, H3SiSiCl3, H3SiSiHCl2, and H2SiSiH2Cl with HAlMe2. For Cl3SiSiCl3 which is the best suited test system, we were not able to locate all structures (ground states and transition states) on the energy surface. For H3SiSiCl3, H3SiSiHCl2, and H2SiSiH2Cl the reaction path via intermediate/transition state A did not show any selectivity in terms of calculated activation energies ((1) reduction step: ΔΔG0AE = 8.6 kcal mol−1; (2) reduction step: ΔΔG0AE = 9.6 kcal mol−1; (3) reduction step: ΔΔG0AE = 11.6 kcal mol−1). In contrast, the calculated activation energies would favor a slightly preferred reaction at the higher chlorinated Si atom, contradicting our experimental observations. Thus, we attribute the unexpected selectivity of our hydrogenation reactions in non-coordinating solvents more to a steric than to an electronic effect. We reasoned that due to the presence of the sterically demanding iBu-groups in transition state A further substitution preferably occurs at the sterically less encumbered Si-center, which is the Si atom bearing already one or two hydrogens leaving the residual SiCl3 groups rather untouched. This steric effect more or less outperforms the electronic influence of the Cl substituents mentioned above leading to the predominant formation of products 4–7 containing exclusively Cl3Si- and H3Si-moieties. Further support for this picture was gained from the observation that the reduction of 7 with iBu2AlH in diethyl ether does not show any selectivity. When 7 was reacted with substoichiometric amounts of iBu2AlH in diethyl ether solution only small amounts of neopentasilane 1 were formed along with insoluble polymeric material and SiH4. In this case the hydrogen atom in iBu2AlH–Et2O containing formally tetravalent aluminum is far more hydridic and nucleophilic and the reaction proceeds via a SN2 mechanism.16b As a consequence the reduction is accompanied by Si–Si bond scission and redistribution reactions and occurs rather unselectively just as found for the hydrogenation of chlorosilanes with LiAlH4 in diethyl ether.
In conclusion, we have introduced two new innovative strategies for the synthesis of chlorohydrooligosilanes in gram quantities. In particular, it has been discovered that substoichiometric amounts of neat iBu2AlH react with perchlorooligosilanes, resulting in the formation of products containing exclusively Cl3Si- and H3Si-moieties. Si–Si bond scission and oligomerization reactions do not occur at all which contrasts the behavior of the more common hydrogenation reagent LiAlH4/diethyl ether described in the literature. Different reaction mechanisms (SN2–Si and SNi–Si) operative in polar and unpolar environments could be made responsible for this unexpected discrepancy. The target molecules of this study are of particular importance for potential applications as alternative precursors for the deposition of hyperpure polycrystalline silicon. Corresponding experiments are currently underway in our laboratories.
This work was supported via a government financed project by the “Austrian Klima and Energiefonds” in the framework of the Austrian energy research project 2015 (FFG Project No. 858491 “Liquid Silicon 2.0”) and the NRW Ministry of Economic Affairs of the state North Rhine-Westphalia (funding code W49, “Liquid Silicon 2.0”). We gratefully acknowledge additional financial support from the Evonik Creavis GmbH.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Research Report on Global and China's Polysilicon Industries, 2019–2023, available athttp://www.researchandmarkets.com.
- (a) F. Chigondo, Silicon, 2018, 10, 789–798 CrossRef CAS; (b) S. Yadav, K. Chattopadhyay and C. V. Singh, Renewable Sustainable Energy Rev., 2017, 78, 1288–1314 CrossRef CAS.
- (a) W. O. Filtvedt, A. Holt, P. A. Ramachandran and M. C. Melaaen, Sol. Energy Mater. Sol. Cells, 2012, 107, 188–200 CrossRef CAS; (b) G. Bye and B. Ceccaroli, Sol. Energy Mater. Sol. Cells, 2014, 130, 634–646 CrossRef CAS; (c) K. Yasuda, K. Morita and T. H. Okabe, Energy Technol., 2014, 2, 141–154 CrossRef CAS.
- (a) M. T. Swihart and R. W. Carr, J. Phys. Chem. A, 1997, 101, 7434–7445 CrossRef CAS; (b) M. T. Swihart and R. W. Carr, J. Phys. Chem. A, 1998, 102, 785–792 CrossRef CAS; (c) M. T. Swihart and R. W. Carr, J. Phys. Chem. A, 1998, 102, 1542–1549 CrossRef CAS.
- S. Ravasio, M. Masi and C. Cavallotti, J. Phys. Chem. A, 2013, 117, 5221–5231 CrossRef CAS.
- X. Meng, H. S. Kim, A. T. Lucero, S. M. Hwang, J. S. Lee, Y.-C. Byun, J. Kim, B. K. Hwang, X. Zhou, J. Young and M. Telgenhoff, ACS Appl. Mater. Interfaces, 2018, 10, 14116–14123 CrossRef CAS.
- (a) M. Abedini, C. H. van Dyke and A. G. MacDiarmid, J. Inorg. Nucl. Chem., 1963, 25, 307–309 CrossRef CAS; (b) R. P. Hollandsworth and M. A. Ring, Inorg. Chem., 1968, 7, 1635–1637 CrossRef CAS; (c) J. E. Drake and N. Goddard, J. Chem. Soc. A, 1970, 2587 RSC; (d) F. Fehér, P. Plichta and R. Guillery, Chem. Ber., 1970, 103, 3028–3033 CrossRef; (e) J. E. Bentham, S. Cradock and E. A. V. Ebsworth, Inorg. Nucl. Chem. Lett., 1971, 7, 1077–1079 CrossRef CAS; (f) F. Feher, P. Plichta and R. Guillery, Inorg. Chem., 1971, 10, 606–608 CrossRef CAS; (g) T. C. Geisler, C. G. Cooper and A. D. Norman, Inorg. Chem., 1972, 11, 1710–1713 CrossRef CAS; (h) N. S. Hosmane, Inorg. Nucl. Chem. Lett., 1974, 10, 1077–1079 CrossRef CAS; (i) F. Fehér and F. Ocklenburg, Z. Anorg. Allg. Chem., 1984, 515, 36–40 CrossRef; (j) J. E. Drake, N. Goddard and N. P. C. Westwood, J. Chem. Soc. A, 1971, 3305–3308 RSC.
- U. Herzog, G. Roewer and U. Pätzold, J. Organomet. Chem., 1995, 494, 143–147 CrossRef CAS.
- U. Herzog and G. Roewer, J. Organomet. Chem., 1997, 544, 217–223 CrossRef CAS.
- (a) K. Hassler and M. Pöschl, J. Organomet. Chem., 1990, 398, 225–227 CrossRef CAS; (b) K. Hassler and W. Köll, J. Organomet. Chem., 1997, 540, 113–118 CrossRef CAS; (c) H. Stüger, J. Organomet. Chem., 1992, 433, 11–19 CrossRef; (d) H. Stüger, J. Organomet. Chem., 1993, 458, 1–7 CrossRef; (e) H. Stüger and P. Lassacher, J. Organomet. Chem., 1993, 450, 79–84 CrossRef.
- H. Stueger, T. Mitterfellner, R. Fischer, C. Walkner, M. Patz and S. Wieber, Inorg. Chem., 2012, 51, 6173–6179 CrossRef CAS.
- (a) H. Stueger, T. Mitterfellner, R. Fischer, C. Walkner, M. Patz and S. Wieber, Chem. – Eur. J., 2012, 18, 7662–7664 CrossRef CAS; (b) J. P. Cannady and X. Zhou, WO2008/051328, 2008; (c) V. Christopoulos, M. Rotzinger, M. Gerwig, J. Seidel, E. Kroke, M. Holthausen, O. Wunnicke, A. Torvisco, R. Fischer, M. Haas and H. Stueger, Inorg. Chem., 2019, 58, 8820–8828 CrossRef CAS.
- (a) F. Porrati, B. Kämpken, A. Terfort and M. Huth, J. Appl. Phys., 2013, 113, 53707 CrossRef; (b) F. Porrati, R. Sachser, G. C. Gazzadi, S. Frabboni and M. Huth, J. Appl. Phys., 2016, 119, 234306 CrossRef; (c) M. Winhold, C. H. Schwalb, F. Porrati, R. Sachser, A. S. Frangakis, B. Kämpken, A. Terfort, N. Auner and M. Huth, ACS Nano, 2011, 5, 9675–9681 CrossRef CAS; (d) T. Bronger, P. H. Wöbkenberg, J. Wördenweber, S. Muthmann, U. W. Paetzold, V. Smirnov, S. Traut, Ü. Dagkaldiran, S. Wieber, M. Cölle, A. Prodi-Schwab, O. Wunnicke, M. Patz, M. Trocha, U. Rau and R. Carius, Adv. Energy Mater., 2014, 4, 1301871 CrossRef.
- M. Haas, V. Christopoulos, J. Radebner, M. Holthausen, T. Lainer, L. Schuh, H. Fitzek, G. Kothleitner, A. Torvisco, R. Fischer, O. Wunnicke and H. Stueger, Angew. Chem., 2017, 129, 14259–14262 CrossRef.
- Compare. (a) I. Fleming, Comprehensive organic chemistry, in The synthesis and reactions of organic compounds, ed. D. Neville Jones, D. H. R. Barton and D. N. Jones, Pergamon Press, Oxford, 1st edn, 1979, pp. 541–686 Search PubMed; (b) A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der anorganischen Chemie, de Gruyter, Berlin, 101st edn, 1995, p. 908 Search PubMed.
- (a) L. H. Sommer, J. McLick and C. M. Golino, J. Am. Chem. Soc., 1972, 94, 669–670 CrossRef CAS; (b) L. H. Sommer, C. M. Golino, D. N. Roark and R. D. Bush, J. Organomet. Chem., 1973, 49, C3–C5 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, and experimental procedures. See DOI: 10.1039/d0cc06506d |
| This journal is © The Royal Society of Chemistry 2020 |