
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A small-molecule probe for monitoring binding to prolyl hydroxylase domain 2 by fluorescence polarisation†
Zhihong
Li
a,
Shuai
Zhen
a,
Kaijun
Su
a,
Anthony
Tumber
b,
Quanwei
Yu
a,
Ying
Dong
a,
Michael
McDonough
 b,
Christopher J.
Schofield
*b and
Xiaojin
Zhang
*ab
b,
Christopher J.
Schofield
*b and
Xiaojin
Zhang
*ab
aLaboratory of Drug Design and Discovery, Department of Chemistry, China Pharmaceutical University, Nanjing 211198, China. E-mail: zxj@cpu.edu.cn
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk
First published on 23rd October 2020
Abstract
Inhibition of the dioxygen sensing hypoxia-inducible factor prolyl hydroxylases has potential therapeutic benefit for treatment of diseases, including anaemia. We describe the discovery of a small-molecule probe useful for monitoring binding to human prolyl hydroxylase domain 2 (PHD2) via fluorescence polarisation. The assay is suitable for high-throughput screening of PHD inhibitors with both weak and strong affinities, as shown by work with clinically used inhibitors and naturally occurring PHD inhibitors.
Hypoxia in animals is associated with diseases including anaemia and cancer. The α,β-heterodimeric hypoxia-inducible transcription factors (HIFs) are of central importance in the chronic response to hypoxia.1 HIF-α levels are regulated in a dioxygen availability limited manner by the C-4 hydroxylation of conserved prolyl-residues, a post-translational modification that signals for HIF-α degradation via the ubiquitin-proteasome machinery.2 In normoxia, HIF-α levels rise, it dimerises with HIF-β, and HIF-mediated transcription is increased.2 The Fe(II) and 2-oxoglutarate (2OG) dependent HIF-α prolyl hydroxylases (PHD1–3 in humans) (Fig. 1A) act as hypoxia sensors for the HIF system.3 Since HIF targets induce erythropoietin and vascular endothelial growth factor, PHD inhibition is of therapeutic interest;4 PHD inhibitors that are 2OG competitors have recently been approved for the treatment of anaemia in chronic kidney disease and others are in clinical trials.5 Reduced activity of PHD and other 2OG oxygenases as a consequence of metabolic reprogramming in cancer cells, via PHD inhibition due to elevated succinate, fumarate, and/or 2-hydroxyglutarate levels, is proposed to be important in disease progression.6 There is thus a need for efficient and accurate assays for monitoring both the catalytic activity of the PHDs and the affinity of natural compounds to their active sites.
 | ||
| Fig. 1 Structure-based design of an affinity-based small-molecule FP probe for PHD2 with its Fe(II) cofactor. (A) PHD catalysed HIF-α prolyl hydroxylation. (B) PHD2 inhibitor 1. (C) Proposed binding mode of 1 based on a reported structure (PDB: 2G19);15 (D) principle of the affinity-based FP probe. | ||
We and others have reported various assays for measuring PHD catalysis,7–11 including by monitoring 2OG consumption7 and by monitoring substrate depletion/product formation by mass spectrometry (MS)/nuclear magnetic resonance (NMR),8,9 or antibody based methods.10 Whilst these assays can be useful for identifying potent PHD inhibitors, they do not provide information on binding constants, in particular to the biologically relevant PHD–Fe complexes. This is important because substantial conformational changes occur during PHD catalysis and, likely, during the binding of some inhibitors.12 Turnover assays are also not ideal for monitoring the binding of compounds with weak affinities, e.g., fragments and natural metabolites, including tricarboxylic acid (TCA) cycle related compounds.13 Here we report the discovery of a fluorescence polarisation (FP) utilising affinity assay for PHD2; importantly, the assay employs the metal Fe(II) cofactor and is suitable for screening metal-binding fragment-like inhibitors.
The design of the probe was based on the recently reported triazole containing PHD inhibitor 1 (Fig. 1B), which is a potent PHD2 inhibitor with an IC50 value of 78 nM as assessed by an FITC-labelled HIF-1α peptide probe-based FP binding assay, both against isolated PHD2 and in cells, and which manifests a promising safety profile.14 Modelling studies based on crystal structures of PHD2 in complex with other inhibitors, imply that 1 chelates the active site Fe(II) in a bidentate manner and its glycine side chain occupies the region normally occupied by the methylene and C-5 carboxylate of 2OG (Fig. S1, ESI†).15 Notably, these studies implied that the meta-position of the chlorophenyl group extends towards the outside of the active site (Fig. 1C), suggesting that functionalisation at this position with a fluorescent reporter group, such as fluorescein isothiocyanate (FITC) could be achieved without substantially perturbing active site binding (Fig. 1D).
Probe 12 was synthesised from phenol derivative 2 (Fig. 2), via3, which was obtained by alkylation of 2 with 1,2-dibromoethane. Methyl ester hydrolysis of 3 yielded 4, which was condensed with N-Boc-1,6-hexanediamine (5) to yield amide 6, which was reacted with NaN3 to give 7. Microwave promoted Huisgen click reaction between 7 and 814 gave triazole 9, which was hydrolysed by base to give 10. Boc-deprotection (CF3CO2H) gave 11, which was derivatised with the reporter group (FITC), then subjected to ester hydrolysis, producing the designed functional PHD2 probe 12.
 | ||
| Fig. 2 Synthesis of PHD probe 12. Reagents and conditions: (a) 1,2-dibromoethane, K2CO3, CH3CN, 60 °C, 9 h, 62%; (b) 30% KOH, 2 h, 91%; (c) EDCI, HOBT, TEA, CH2Cl2, rt, 5 h, 54%; (d) NaN3, DMF, 100 °C, 6 h, 91%; (e) TBAF, DIPEA, CuI, MeOH, 80 °C, 4 h, 59%; (f) LiOH, H2O, THF, 30 °C, 92%; (g) CF3COOH, CH2Cl2, rt, overnight, 56%; (h) FITC, HBTU, DIPEA, DMF, rt; then LiOH, H2O, THF, 30 °C, 45% (structure of probe 12 see Fig. 3A). | ||
To investigate the effects of introducing the linker and bulky FITC group on the probe's ability to bind PHD2, the affinity of 10 with PHD2 was evaluated using our reported FP assay, which manifested an IC50 of 310.6 ± 9.7 nM (Fig. S2, ESI†). This observation suggests that the introduction of the linker and FITC group does not ablate the strong affinity of the active site binding elements of the probe, though its affinity is reduced somewhat. Encouraged by this result, we then investigated the biological applications of probe 12. First, a solid phase extraction (SPE)-MS coupled assay16 was performed to evaluate the potency of probe 12 for PHD2 inhibition. The resultant IC50 was 166 ± 3 nM, which compares favourably with a PHD inhibitor approved for clinical use to treat anaemia associated with chronic kidney disease (CKD)17 (FG-4592, IC50: 2587 ± 20 nM) (Fig. 3B). The direct binding affinity of probe 12 to PHD2 was determined by monitoring the FP signals; the resulting EC50 value of probe 12 that binds to PHD2 is 27.4 ± 1.2 nM (Fig. 3C).
 | ||
| Fig. 3 (A) Structure of probe 12. (B) Dose–response inhibition curves of probe 12 and FG-4592 based on the SPE MS assay;16 (C) dose–response binding curve of probe 12 based on the titration FP assay (no additional iron ion and 2OG were added). See ESI† for details. | ||
The selectivity of probe 12 towards selected other human 2OG oxygenases, such as factor inhibiting HIF (FIH) and JmjC histone Nε-methyl lysine demethylases (KDMs), was then evaluated using established assays (Table 1). The results indicated that probe 12 is highly selective for PHD2 over the tested 2OG dependent KDMs. Probe 12 was selective for PHD2 over FIH (which regulates the transcriptional activity of HIF), though to a lesser extent.
We then developed a small-molecule fluorescent probe-based FP assay for PHD2. The influence of the polarity of the medium on the fluorescence intensity of probe 12 was investigated by adding dioxane or DMSO in PBS buffer measured at a λmax of 520 nm, respectively (Fig. S3, ESI†). The excitation and emission spectra of probe 12 are characterised by maxima λ = 505 nm (excitation) and 520 nm (emission) (Fig. 4). The results indicated that probe 12 has acceptable spectroscopic properties in the presence of dioxane or DMSO in buffer when measured at a λmax of 520 nm (Fig. S3, ESI†). Thus, probe 12 was considered suitable for further development as a tool for studies on PHD2.
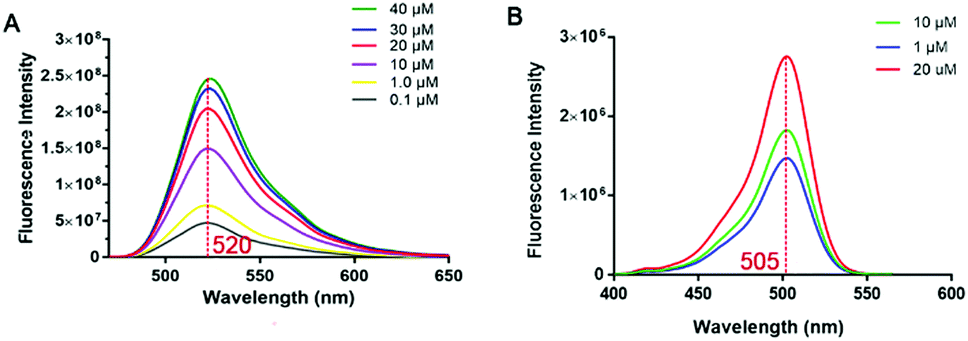 | ||
| Fig. 4 Spectroscopic properties of probe 12. (A) Emission spectra of probe 12. (B) Excitation spectra of probe 12. | ||
We optimised use of probe 12 for efficient affinity-based FP method for quantitative screening of PHD2 inhibitors. The developed method does not require the addition of 2OG and can employ Fe(II) that copurifies with PHD2,20 though apo-protein and added metal ions can also be used (Fig. S4 and S5, ESI†). The assay employs low concentrations of probe 12 (30 nM) and PHD2 (20 nM) (Fig. 5A), the latter is a level much lower than most previously reported assays, e.g. employing matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) (50 μM),8 NMR (10 μM),9 or peptide-based FP assays (100 nM).11 We investigated the dependency of the binding affinity on factors including pH (range 4–10), DMSO concentration, and incubation time (Fig. S6–S9, ESI†). The assay is robust with Z′ values ≥0.80 (Fig. S9, ESI†). Once PHD2 and probe 12 had attained equilibrium (30 min), the complex was stable over 24 h (Fig. S7, ESI†). The binding affinity was reasonably stable in the presence of up to 16% (v/v) DMSO (Fig. S8, ESI†). The new assay complements turnover based methods and is more efficient, simpler, and lower cost than previous methods (Fig. 5B).
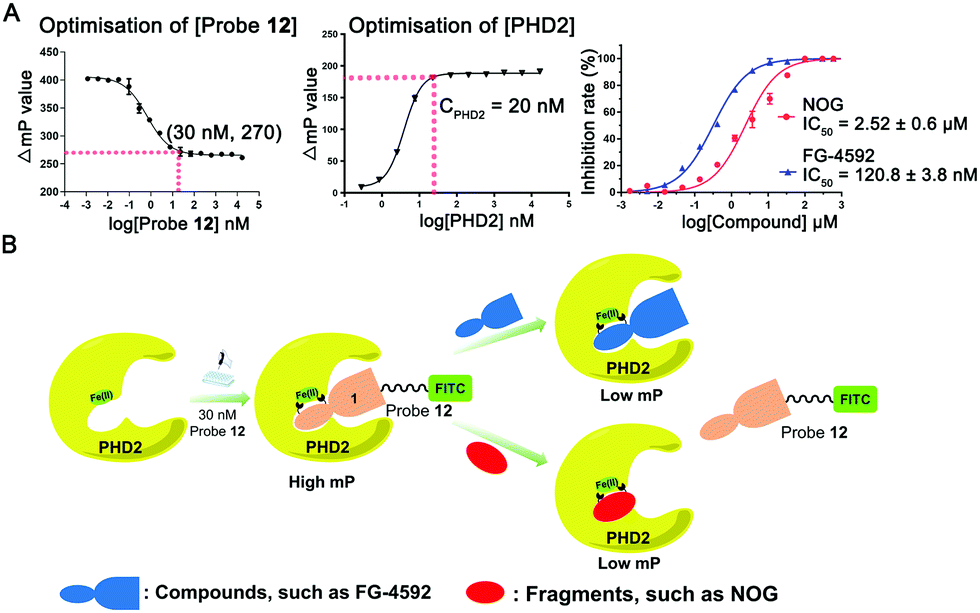 | ||
| Fig. 5 (A) Optimisation of the concentration of probe 12 and PHD2 and inhibition by FG-4592 and NOG; (B) schematic representation of FP assays used to investigate the binding of probe 12 and PHD2 and the displacement of probe 12 by PHD2 inhibitors. | ||
We then validated the assay by evaluating the affinities of the clinical inhibitors FG-4592 (Roxadustat), AKB-6548 (Vadadustat), and 1 for PHD2. The IC50 values of FG-4592, AKB-6548, and 1 for inhibition of the PHD2/probe 12 interaction were 120.8 ± 3.8 nM, 215.1 ± 2.1 nM, and 21.5 ± 2.3 nM, respectively. These values are in accord with the IC50 values determined by the HIF-1α peptide-based FP (FG-4592, 591.4 nM; AKB-6548, 608.7 nM; 1, 77.7 nM)11 and SPE MS substrate turnover (FG-4592, 2.587 μM; 1, 0.276 μM) assays (Fig. 6). Though there are variations in the absolute IC50 values derived from the probe 12-based FP assay, the HIF-1α peptide-based FP assay, or SPE MS substrate turnover assays, due to the different experimental conditions,21 the trends of the inhibitory activity towards PHD2 are consistent for all of these.
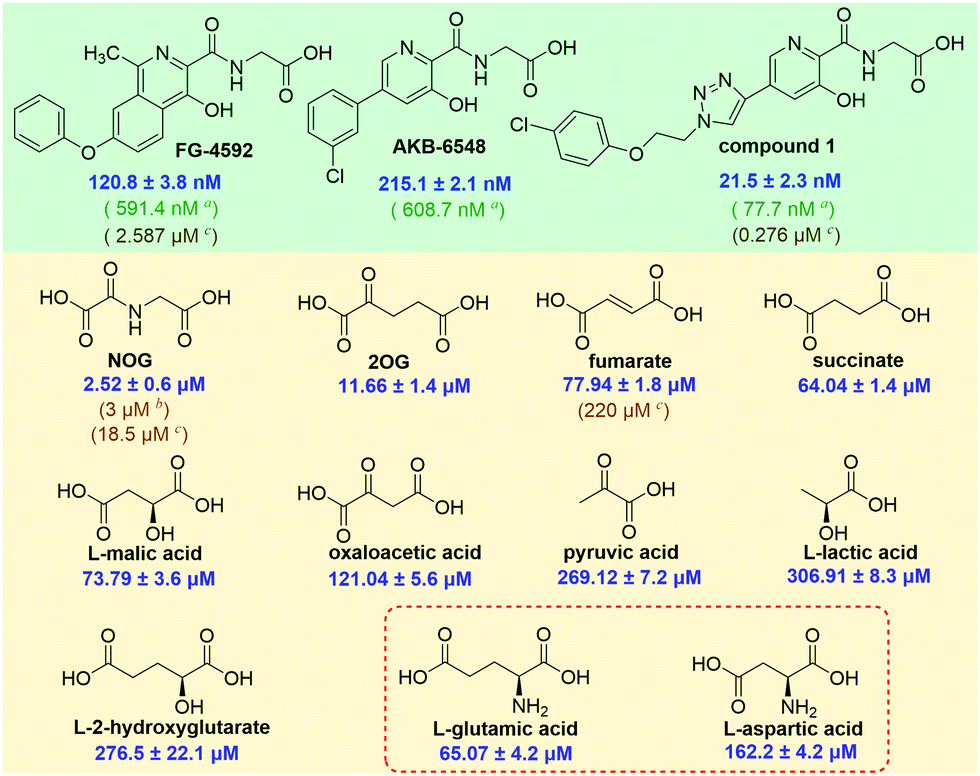 | ||
Fig. 6 IC50 values of these representative compounds (a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) HIF-1α peptide-based FP assay; bNMR assay; cSPE MS assay). HIF-1α peptide-based FP assay; bNMR assay; cSPE MS assay). | ||
Thus, we then used probe 12 to evaluate the binding affinities of naturally occurring 2OG analogues and PHD2 (Fig. 5B). The assay reveals that N-oxalylglycine (NOG), a plant metabolite and 2OG isostere,18 inhibits the PHD2/probe 12 interaction with an IC50 of 2.52 ± 0.6 μM. This value is similar to the IC50 (3 ± 1 μM) determined by an NMR binding assay,9 though lower than that obtained by MS based turnover assay (IC50 of 18.5 μM).8 It is proposed that in cells PHD activity may be regulated by TCA cycle intermediates and related compounds,19,20,22 thus we used our assay to investigate affinities of such compounds for PHD2 (Fig. 6 and Table S1, ESI†). Consistent with prior work using NMR-based and MS-based assays,9,13 fumarate, and succinate bind weakly to PHD2 with IC50 values of 77.94 ± 1.8 μM and 64.0 ± 1.4 μM, respectively; consistent with prior studies 2-hydroxylgutarate also bound weakly (Fig. 6).
Interestingly, the assay revealed that dicarboxylic amino-acids including aspartate (IC50: 162.2 ± 4.2 μM) and glutamate (IC50: 65.07 ± 4.2 μM) bind PHD2, albeit weakly (Fig. 6). The concentrations of some amino acids, are high in the cytosol; this is particularly true for glutamate where concentrations of ≥10 mM are reported in some cells.23N-Acylated derivatives of Glu, are reported as PHD inhibitors,24 but to our knowledge there are no reports of Glu itself binding to the PHDs. There is thus a possibility of crosstalk between amino acid metabolism and the HIF pathway/2OG oxygenase catalysis in cells. If indeed PHD activity in cells is mediated by glutamate, there are implications for intracellular inhibition of the PHDs by other small-molecules, both by drugs and TCA metabolites and the related oncometabolite 2-hydroxyglutarate.6,25
The overall results demonstrate that the small-molecule probe 12-based FP assay is an efficient and cost-effective method for analysis of both strong and weakly binding fragment-like PHD2 inhibitors. The assay complements reported fluorescence based assays for 2OG oxygenases,26 requires small amounts of reagents compared to turnover-assays8–10 and is suited to HTS application. Probe 12 binds well to FIH (Table 1); thus there is scope for its further development. Probe 12 did not manifest cytotoxicity in HEK293, Hep3B, and L02 cells (Fig. S10, ESI†), so may also be useful in exploring PHD biology and inhibition in cells.
In comparison to our reported affinity-based assay for PHD2 using a FITC-labelled HIF-1α (556–574) peptide as the FP probe,11 the probe 12-based FP assay has the advantage that it works with Fe(II); the HIF-1α peptide-based FP assay employs Mn(II) instead of Fe(II), because Fe(II)-complexed PHD2 hydroxylates the peptide probe causing it to lose affinity (Fig. S5B, ESI†).11 Additionally, given that the HIF peptide probe does not occupy key regions of the active site, e.g. the 2OG binding pocket, the HIF-1α peptide-based FP assay fails to identify certain types of inhibitor, including fragment-like compounds, e.g. NOG (Fig. S11, ESI†), a problem overcome by the probe 12-based FP assay. The combined use of the two assays can provide information on whether inhibitors compete with HIF-α and/or 2OG. The assay was validated by work with clinically used inhibitors and naturally occurring fragment-like PHD inhibitors, such as TCA cycle intermediates and amino acids, the results of which are being followed up. We envision future applications of probe 12 in studying the biological roles of the PHDs and crosstalk between the HIF pathway, the TCA cycle, and amino acid metabolism.
This work was supported by grants from the National Natural Science Foundation of China (grant code: 81973173), Jiangsu Province Funds for Excellent Young Scientists (grant code: BK20170088), the Six Talent Peaks Project (grand code: YY-023) of Jiangsu Province, the Wellcome Trust (grant code: 106244/Z/14/Z) and Cancer Research UK (grant code: C9047/A24759).
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- R. Chowdhury, A. Hardy and C. J. Schofield, Chem. Soc. Rev., 2008, 37, 1308–1319 RSC.
- M. H. Rabinowitz, J. Med. Chem., 2013, 56, 9369–9402 CrossRef CAS.
- C. J. Schofield and P. J. Ratcliffe, Nat. Rev. Mol. Cell Biol., 2004, 5, 343–354 CrossRef CAS.
- A. A. Joharapurkar, V. B. Pandya, V. J. Patel, R. C. Desai and M. R. Jain, J. Med. Chem., 2018, 61, 6964–6982 CrossRef CAS.
- Z. Li, Q. You and X. Zhang, J. Med. Chem., 2019, 62, 5725–5749 CrossRef CAS.
- H. Tarhonskaya, A. M. Rydzik, I. K. H. Leung, N. D. Loik, M. C. Chan, A. Kawamura, J. S. O. McCullagh, T. D. W. Claridge, E. Flashman and C. J. Schofield, Nat. Commun., 2014, 5, 4423 CrossRef.
- L. A. McNeill, L. Bethge, K. S. Hewitson and C. J. Schofield, Anal. Biochem., 2005, 336, 125–131 CrossRef CAS.
- C. J. Stubbs, C. Loenarz, J. Mecinovic, K. K. Yeoh, N. Hindley, B. M. Lienard, F. Sobott, C. J. Schofield and E. Flashman, J. Med. Chem., 2009, 52, 2799–2805 CrossRef CAS.
- I. K. H. Leung, M. Demetriades, A. P. Hardy, C. Lejeune, T. J. Smart, A. Szollossi, A. Kawamura, C. J. Schofield and T. D. W. Claridge, J. Med. Chem., 2013, 56, 547–555 CrossRef CAS.
- R. Chowdhury, J. I. Candela-Lena, M. C. Chan, D. J. Greenald, K. K. Yeoh, Y. M. Tian, M. A. McDonough, A. Tumber, N. R. Rose, A. Conejo-Garcia, M. Demetriades, S. Mathavan, A. Kawamura, M. K. Lee, F. van Eeden, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, ACS Chem. Biol., 2013, 8, 1488–1496 CrossRef CAS.
- Y. Lei, T. Hu, X. Wu, Y. Wu, Q. Bao, L. Zhang, H. Xia, H. Sun, Q. You and X. Zhang, ACS Med. Chem. Lett., 2015, 6, 1236–1240 CrossRef CAS.
- C. Domene, C. Jorgensen and C. J. Schofield, J. Am. Chem. Soc., 2020, 142, 2253–2263 CrossRef CAS.
- K. S. Hewitson, B. M. Lienard, M. A. McDonough, I. J. Clifton, D. Butler, A. S. Soares, N. J. Oldham, L. A. McNeill and C. J. Schofield, J. Biol. Chem., 2007, 282, 3293–3301 CrossRef CAS.
- Y. Wu, Z. Jiang, Z. Li, J. Gu, Q. You and X. Zhang, J. Med. Chem., 2018, 61, 5332–5349 CrossRef CAS.
- M. A. McDonough, V. Li, E. Flashman, R. Chowdhury, C. Mohr, B. M. Lienard, J. Zondlo, N. J. Oldham, I. J. Clifton, J. Lewis, L. A. McNeill, R. J. Kurzeja, K. S. Hewitson, E. Yang, S. Jordan, R. S. Syed and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9814–9819 CrossRef CAS.
- J. P. Holt-Martyn, R. Chowdhury, A. Tumber, T.-L. Yeh, M. I. Abboud, K. Lippl, C. T. Lohans, G. W. Langley, W. Figg Jr., M. A. McDonough, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, ChemMedChem, 2020, 15, 270–273 CrossRef CAS.
- S. Dhillon, Drugs, 2019, 79, 563–572 CrossRef CAS.
- K. Al-Qahtani, B. Jabeen, R. Sekirnik, N. Riaz, T. D. W. Claridge, C. J. Schofield and J. S. O. McCullagh, Phytochemistry, 2015, 117, 456–461 CrossRef CAS.
- M. A. Selak, S. M. Armour, E. D. MacKenzie, H. Boulahbel, D. G. Watson, K. D. Mansfield, Y. Pan, M. C. Simon, C. B. Thompson and E. Gottlieb, Cancer Cell, 2005, 7, 77–85 CrossRef CAS.
- R. V. Duran, E. D. MacKenzie, H. Boulahbel, C. Frezza, L. Heiserich, S. Tardito, O. Bussolati, S. Rocha, M. N. Hall and E. Gottlieb, Oncogene, 2013, 32, 4549–4556 CrossRef CAS.
- T.-L. Yeh, T. M. Leissing, M. I. Abboud, C. C. Thinnes, O. Atasoylu, J. P. Holt-Martyn, D. Zhang, A. Tumber, K. Lippl, C. T. Lohans, I. K. H. Leung, H. Morcrette, I. J. Clifton, T. D. W. Claridge, A. Kawamura, E. Flashman, X. Lu, P. J. Ratcliffe, R. Chowdhury, C. W. Pugh and C. J. Schofield, Chem. Sci., 2017, 8, 7651–7668 RSC.
- M. I. Abboud, T. E. McAllister, I. K. H. Leung, R. Chowdhury, C. Jorgensen, C. Domene, J. Mecinović, K. Lippl, R. L. Hancock, R. J. Hopkinson, A. Kawamura, T. D. W. Claridge and C. J. Schofield, Chem. Commun., 2018, 54, 3130–3133 RSC.
- A. J. L. Cooper and T. M. Jeitner, Biomolecules, 2016, 6, 16 CrossRef.
- D. R. Mole, I. Schlemminger, L. A. McNeill, K. S. Hewitson, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield., Bioorg. Med. Chem. Lett., 2003, 13, 2677–2680 CrossRef CAS.
- P. Koivunen, S. Lee, C. G. Duncan, G. Lopez, G. Lu, S. Ramkissoon, J. A. Losman, P. Joensuu, U. Bergmann, S. Gross, J. Travins, S. Weiss, R. Looper, K. L. Ligon, R. G. W. Verhaak, H. Yan and W. G. Kaelin Jr, Nature, 2012, 483, 484–488 CrossRef CAS.
- X. Luo, Y. Liu, S. Kubicek, J. Myllyharju, A. Tumber, S. Ng, K. H. Che, J. Podoll, T. D. Heightman, U. Oppermann, S. L. Schreiber and X. Wang, J. Am. Chem. Soc., 2011, 133, 9451–9456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and additional experiments. See DOI: 10.1039/d0cc06353c |
| This journal is © The Royal Society of Chemistry 2020 |