
Unravelling the local structure of catalytic Fe-oxo clusters stabilized on the MOF-808 metal organic-framework†
Celia
Castillo-Blas
 a,
Ignacio
Romero-Muñiz
a,
Andreas
Mavrandonakis
b,
Laura
Simonelli
c and
Ana E.
Platero-Prats
*ad
a,
Ignacio
Romero-Muñiz
a,
Andreas
Mavrandonakis
b,
Laura
Simonelli
c and
Ana E.
Platero-Prats
*ad
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Campus de Cantoblanco, Madrid 28049, Spain. E-mail: ana.platero@uam.es
bElectrochemical Processes Unit, IMDEA Energy, Avenida Ramón de la Sagra 3, Móstoles 28935, Madrid, Spain
cCLAESS beamline, ALBA Synchrotron, Cerdanyola del Vallès 08290, Spain
dCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049, Campus de Cantoblanco, Madrid 28049, Spain
First published on 23rd November 2020
Abstract
Stabilizing catalytic iron-oxo-clusters within nanoporous metal–organic frameworks (MOFs) is a powerful strategy to prepare new active materials for the degradation of toxic chemicals, such as bisphenol A. Herein, we combine pair distribution function analysis of total X-ray scattering data and X-ray absorption spectroscopy, with computational modelling to understand the local structural nature of added redox-active iron-oxo clusters bridging neighbouring zirconia-nodes within MOF-808.
Metal–Organic Frameworks (MOFs) are porous and crystalline materials composed of organic ligands linked by metal-oxo nanoclusters, to give open architectures with large surface areas and pores of different shapes and sizes.1 The exceptional textural and chemical properties of these materials make them excellent candidates for the selective capture and degradation of many types of hazardous chemicals.2,3 In particular, the family of Zr(IV)-MOF materials presents a high thermal and chemical stability in water,4 allowing applicability in the aqueous-based degradation of pesticides, pharmaceuticals and pollutants. The Zr(IV)-MOFs are composed of Zr6O8 clusters, which can be 12-, 8-, or 6-fold connected to carboxylate organic ligands, UiO-type, NU-1000 and MOF-808 being the archetypical ones, respectively.5 MOF-808 is an interesting platform to target liquid-phase applications, including capture and degradation processes.6 Its structure is built from linking unsaturated Zr6O8 clusters by benzene-1,3,5-tricarboxilate (BTC) ligands, to give mesopores of around 20 Å size. Besides its high porosity, MOF-808 has a remarkably highly tunable structure arising from the low saturation of the Zr6O8 clusters. Within the octahedral Zr6O8 clusters, the linking positions around the equatorial plane can serve as scaffolds to insert discrete functional groups, including aminoacids,7 sulfates,8 and metal complexes.9 Furthermore, the presence of available hydroxyl groups within the Zr6O8 clusters represents an excellent platform to further modify the MOF-808 chemistry with transition metals.
Solvothermal incorporation in MOF (SIM) materials has been widely explored to decorate Zr6O8 clusters, by soaking the pristine MOF into a solution containing a target transition metal precursor.10 Thus, a variety of MOF chemical modifications with catalytic transition metal sites have been reported using this method.11 In this regard, exploring the incorporation of a redox-active transition metal, such as iron, would be a promising approach for tailoring MOF catalytic properties towards the oxidative degradation of organic pollutants such as endocrine disruptor chemicals (EDCs). In this context, Fenton reactions catalyzed by iron(II) are a powerful methodology to afford the oxidative degradation of organic pollutants into mainly carbon dioxide and water.12,13 However, acidic conditions are typically required to stabilize iron species in water during catalysis. Therefore, developing catalytic Zr(IV)-MOF materials with intrinsic highly acidic surfaces and containing redox-active iron sites, able to capture and subsequently convert pollutants into non-toxic compounds, would represent an elegant strategy to degrade EDC in water under mild conditions.
In this work, we show the SIM modification of MOF-808 with binuclear iron clusters, active for the selective capture and subsequent catalytic degradation of an EDC compound, such as bisphenol A (BPA) via Fenton reactions. Detailed synchrotron characterization, including pair distribution function (PDF) analysis of X-ray total scattering data and Fe K-edge X-ray absorption spectroscopy (XAS) together with density functional theory (DFT) computational modelling, is applied to understand the local structural nature of the added redox-active iron clusters within MOF-808.
The SIM synthesis of Fe-MOF-808 was performed by immersion of the pristine material, previously activated, in a solution containing FeCl2 in N,N′-dimethylformamide (see the ESI†). To explore the kinetic aspects of the iron incorporation within MOF-808, different Fe-SIM reaction times were tested using two different starting Fe to Zr6 molar ratios. Powder X-ray diffraction (PXRD) data collected on the activated materials showed the presence of the MOF-808 phase for all reaction times. Scanning electron microscopy and energy dispersive X-rays spectroscopy (SEM-EDS) analyses indicated that the incorporation of iron within MOF-808 strongly depends on the Fe-SIM reaction time. The final Fe to Zr6O8 molar ratio determined was ca. 0.5 (i.e. Fe-0.5-MOF-808). Similar behaviour was observed for the synthesis of the high-content Fe-1.2-MOF-808 (S4, ESI†). These results demonstrated that optimal iron MOF modification is obtained at long Fe-SIM reaction times, resulting in materials with enhanced homogeneity in their chemistries and lacking unwanted iron oxide nanoparticles as by-products.
SEM-EDS mapping elemental analyses showed a highly homogeneous distribution of iron along the MOF crystallites for both 0.5- and 1.2-MOF-808 (Fig. 1C). Pawley refinements of the diffraction data corroborated that the average MOF architecture is preserved after Fe-SIM, together with the occurrence of a minor cell expansion and peak broadening compared to the pristine system (Fig. 1D and Table S7.1, ESI†). N2 isotherms revealed an expected decrease in specific surface area associated with the iron incorporation within the MOF-808 structure together with subtle variations in the pore size distribution (Fig. 1A and B and Fig. S4, ESI†). Detailed analysis of the pore volume contributions showed the occurrence of a significant decrease in volume linked to the mesopores of 18 Å (ca. 50%) compared to the minor loss seen for the micropores of 12 Å (Fig. 1B and Fig. S4, ESI†). These results would suggest that the iron sites are located pointing towards the MOF-808 hexagonal channels.
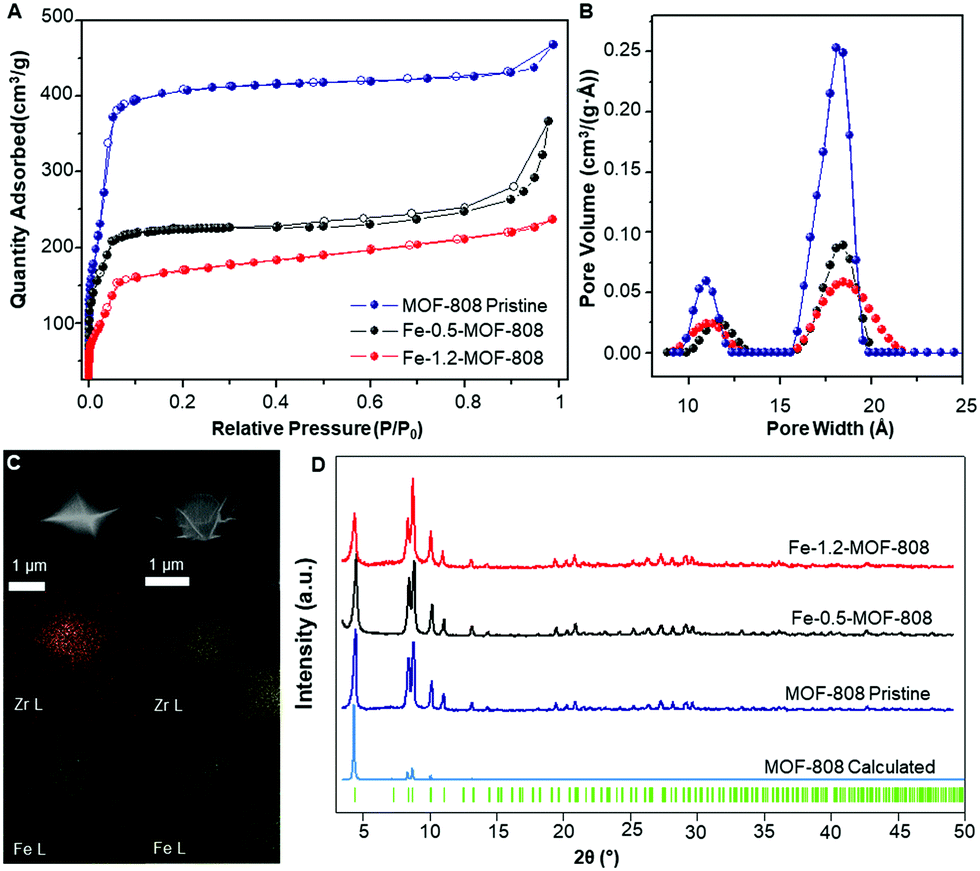 | ||
| Fig. 1 (A) N2 adsorption/desorption isotherms at 77 K showing the decrease in surface area upon iron incorporation. (B) Pore size distribution (PSD) data for pristine and Fe-SIM MOF-808 materials. (C) Mapping of Zr (yellow) and Fe (green) for Fe-0.5-MOF-808 (right) and Fe-1.2-MOF-808 (left). (D) PXRD data of the synthesized materials and their comparison with the calculated data for MOF-808,19 including Bragg positions. | ||
To probe the atomic local structure of the iron sites within MOF-808, we applied PDF analysis of X-ray total scattering data (xPDF). xPDF data collected on the Fe-SIM and pristine MOF-808 materials showed two major contributions at short range, that is, Zr–O (ca. 2.2 Å) and Zr⋯Zr (ca. 3.5 Å) distances, characteristic of the Zr6O8 clusters.14,15 With the purpose of highlighting the subtle contributions in this region that are associated with the iron sites, differential analysis of the xPDF data16 was carried out by comparing the PDF of the pristine material to that of the Fe-MOF-808 systems. d-PDF analysis carried out on the Fe-MOF-808 materials revealed the appearance of new correlations associated with the iron-clusters at ∼1.9–2.0 Å, ∼2.3 Å and ∼3.0 Å (Fig. 2A). The Fe–O distance values are sensitive to the oxidation state of iron,17 being shorter for Fe(III) (∼1.85 Å) compared to Fe(II) (∼2.05 Å). The broad d-PDF signal observed at ∼1.9–2.0 Å indicated the presence of mixed oxidation states for iron within the Fe-MOF-808 systems. In addition, the presence of chloride groups bonded to the iron sites is confirmed by the peak centred at ∼2.3 Å, associated with Fe–Cl bonds. Interestingly, the correlation centred at ∼3.0 Å is characteristic of Fe–O–Fe distances within edge-sharing geometries.18 Additionally, two d-PDF peaks centred at ∼3.3 and ∼3.7 Å are observed, which could be attributed to the Zr⋯Fe correlations. These results indicated the presence of iron-oxo clusters binding the Zr6-nodes within the Fe-MOF-808 materials, where some of the oxygen positions have been replaced by chloride, as suggested by SEM-EDS.
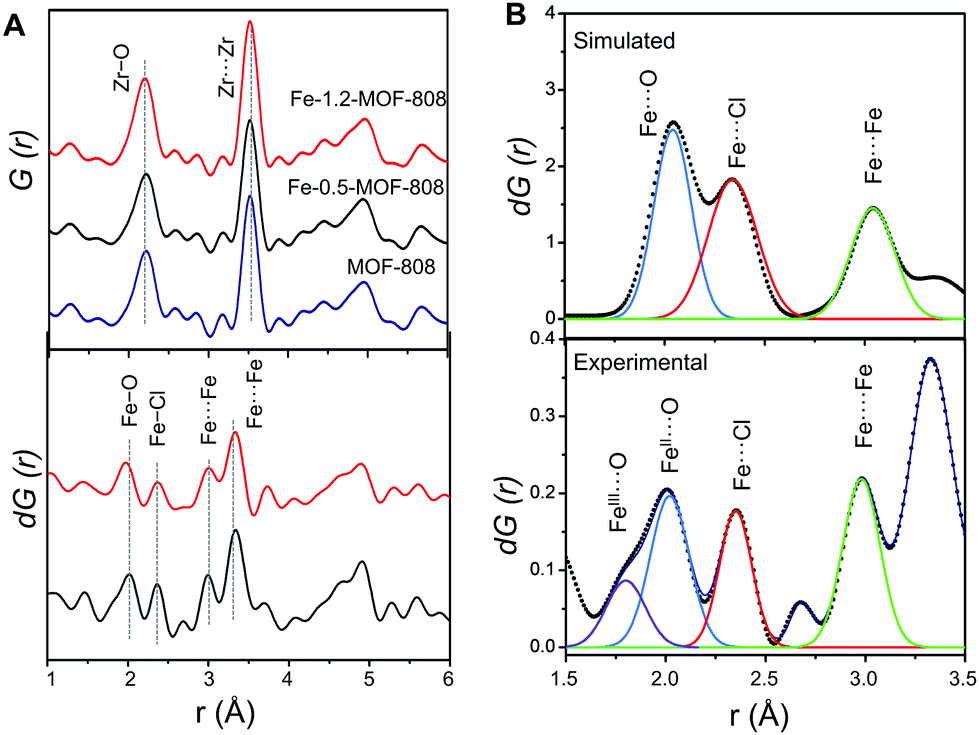 | ||
| Fig. 2 (A) Total PDFs of pristine and Fe-MOF-808 materials (up) and the corresponding d-PDFs for Fe-0.5-MOF-808 and Fe-1.2-MOF-808. dPDF signals were obtained after comparing the PDF signal of pristine MOF-808 to that of Fe-MOF-808 materials, in order to highlight the correlations characteristic of the iron clusters (down). (B) Simulated d-PDF signal of a hypothetical binuclear iron cluster (up) and experimental d-PDF signal of Fe-0.5-MOF-808 (down) fitted to Gaussian curves for quantitative analyses to estimate the cluster size. | ||
Quantitative analysis of the relative d-PDF peak intensities is a powerful strategy to estimate the size of metal-oxo clusters deposited in MOF-808.20 Thus, the signals linked to Fe–O, Fe–Cl and Fe–O–Fe were fitted to Gaussian curves and compared to preliminary models of different iron cluster sizes (i.e. bearing two, three or four iron atoms, Fig. 2B). By comparing the experimental d-PDF peak intensities of the Fe–O,Cl and contributions to simulated values, we hypothesized that Fe-0.5-MOF-808 contains clusters of two iron atoms in distorted tetrahedral environments. Furthermore, the results suggest that the iron sites are bonded to both chlorine and oxygen atoms (including aquo, hydroxo or oxo group) in a 1 to 2 ratio (Fig. S8.3 and Table S8.1, ESI†). For Fe-1.2-MOF-808, quantitative analyses indicated the formation of slightly larger iron clusters compared to Fe-0.5-MOF-808 (Fig. S8.4 and Table S8.2, ESI†), suggesting that the use of high concentrations of the iron precursor would favour aggregation.
To better understand the chemical nature of the binuclear iron clusters within MOF-808, Fe K-edge extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge spectroscopy (XANES) data were collected on the Fe-MOF-808 materials. XANES data showed the occurrence of rising edge and pre-edge features at ca. 7122 and ca. 7114 eV, characteristic of binuclear iron complexes.21 A slight shift of rising-edge position of ca. 1 eV to higher values is observed for Fe-1.2-MOF-808 compared to Fe-0.5-MOF-808, in agreement with the oxidation of Fe2+ to Fe3+ (Fig. 3A). The observed pre-edge signal corresponds to the Fe-1s to Fe-3d electronic transition, and its intensity is indicative of the tetrahedral geometries of the iron sites in both materials.22 This experimental evidence is in agreement with the most energetically favoured geometry compatible with both II/III oxidation states found for iron within these materials. Further analyses of the EXAFS data collected on Fe-0.5-MOF-808 indicated the presence of three main signals at ca. 1.35, ca. 1.81 and 2.58 Å (without phase correction), linked to FeII–O, Fe–Cl (2.25 Å) and Fe⋯Fe distances, respectively (Fig. 3B).23 Interestingly, EXAFS data of Fe-1.2-MOF-808 showed a single contribution at ca. 1.56 Å (without phase correction) linked to FeIII–Cl,O bonds together with a significant increase in the intensity of the signal linked to Fe⋯Fe distances. This evidence corroborates the presence of larger clusters with an oxidized state of iron in Fe-1.2-MOF-808 compared to Fe-0.5-MOF-808, in agreement with the xPDF analyses.
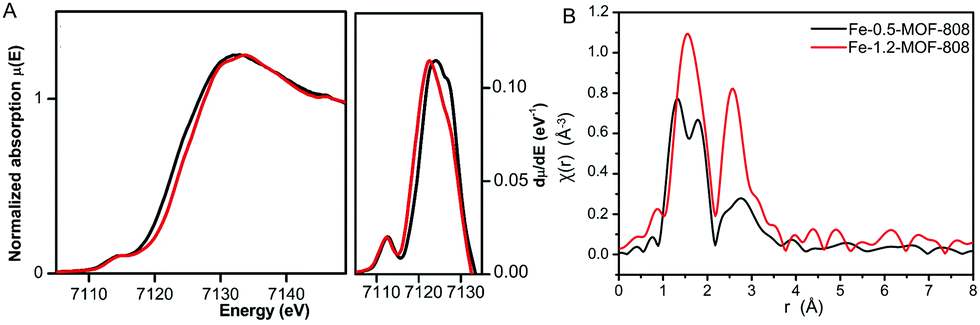 | ||
| Fig. 3 (A) Fe K-edge XANES data of Fe-MOF-808 materials (left) and first derivate analysis (right) showing the rising-edge shift between both materials. (B) The k3-weighted χ(r) Fe K-edge EXAFS spectra of Fe-0.5- and Fe-1.2-MOF-808 systems. | ||
Density Functional Theory (DFT) calculations were performed in order to elucidate the possible conformations of the iron bimetallic core of the Fe-MOF-808. The structural and energetic properties of several binuclear iron-oxo and iron-hydroxo clusters deposited on the nodes of the MOF-808 were modelled. The approach in this study is similar to previous works, where the deposition of small copper-, cobalt- and nickel-(hydro)oxo clusters was investigated on the nodes of the NU-1000.20,24,25 An exhaustive description of the possible iron–(hydro)oxo clusters explored can be found in the ESI,† Section 10. Among the possible structures investigated, two possess structural features that agree with the experimental PDF. Moreover, from a thermodynamic point of view, they are the most stable conformations because of the most negative computed free energies of formation. Our calculations demonstrated that the iron-precursors may react with: (i) two terminal-aqua protons giving rise to the conformation A (bridging site) or (ii) one terminal-aqua proton giving rise to conformation B (or terminal site). In both cases, the bimetallic iron core is deposited as Fe2Cl2(μ2-OH)x(OH)y on the nodes binding through terminal-OH groups within the Zr6O8 units. In one case, the iron-hydroxo cluster bridges two Zr6O8 octahedra in a μ2 mode (conformation A), while in the second case (conformation B), it is deposited on one node in a η2μ2 mode and the cluster is terminated by an additional hydroxo group (Fig. 4). The presence of an intense dPDF signal at ca. 3.3 Å, linked to Fe⋯Fe and Fe⋯Zr distances within conformation A (Fig. 4A), demonstrated the stabilization of the Fe-oxo clusters by bridging two Zr6O8 clusters as main species. Additionally, the occurrence of signals at ca. 3.0 and ca. 3.7 Å, associated with Fe⋯Fe and Fe⋯Zr distances within the conformation B (Fig. 4B), evidenced the deposition of the Fe-oxo clusters within the Zr6O8 cluster through terminal hydroxo groups. Remarkably, these results demonstrated that the Fe-oxo clusters are stabilized by bridging two neighbouring Zr6O8 nodes within MOF-808, as previously identified for other zirconia-based MOF systems.20
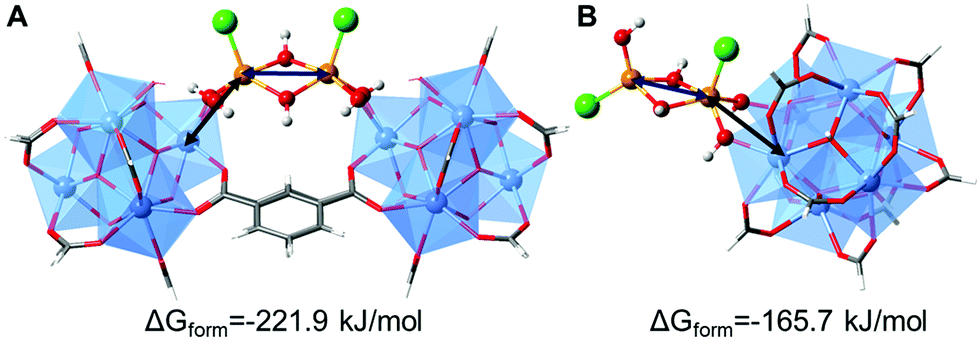 | ||
| Fig. 4 DFT computed structures of possible bimetallic iron–hydroxo clusters deposited on MOF-808, highlighting the most representative Fe⋯Fe and Fe⋯Zr distances observed in the d-PDF data. (A) Iron cluster acts as a bridge between two Zr6O8 nodes. (B) Iron cluster is linked to one Zr6O8 node via a terminal position. The M06-L computed free energies of formation are also given. A more detailed description of the calculations can be found in the ESI† (Section 10). | ||
As a proof of concept, activated Fe-MOF-808 materials were evaluated for the capture and subsequent catalytic degradation of BPA through Fenton reactions. Fe-0.5-MOF-808 was able to capture ca. 50% of BPA after 1 h, contrary to the ca. 11% captured by Fe-1.2-MOF-808 (Fig. S11.1, ESI†). This result is in accordance with the textural properties of the materials (Fig. 1A and B), the MOF with a lower iron content (i.e. Fe-0.5-MOF-808) has larger free space to capture molecules than its analogue with higher iron content (i.e. Fe-1.2-MOF-808). To study the performance of the Fe-MOF-808 materials as catalysts, the degradation of BPA was carried out through a Fenton reaction using H2O2 (see details in the ESI†). The catalytic degradation was compared at 30 and 60 min reaction time (Fig. S11.1, ESI†). Interestingly, the structural differences seen for the two MOFs also play a role in their catalytic performance regarding both the kinetic and the thermodynamic aspects. Thus, Fe-0.5-MOF-808 degraded 26% of the total BPA after 30 minutes, this value being three times better than the activity determined for Fe-1.2-MOF-808. After 1 hour of reaction time, a plateau is reached and ca. 40% of the total BPA is removed by the low Fe-loaded material, this value being remarkably larger than the performance observed for Fe-1.2-MOF-808 containing larger iron-oxo clusters.
In conclusion, we have reported the stabilization of catalytic Fe-oxo clusters within MOF-808 through SIM methods, active in the degradation of BPA via Fenton reactions. Interestingly, synchrotron XAFS and PDF characterization combined with DFT modelling demonstrated the occurrence of Fe-oxo dimers bridging two neighbouring Zr6O8 nodes as the main species. We hypothesize that the terminal Fe-oxo species (conformation B) might be formed under kinetic control at short reaction times, and subsequently react with a terminal hydroxo group from a neighbouring zirconia node giving the formation of the most thermodynamically stable bridging species (conformation A). Further studies are currently under investigation to elucidate the structural mechanism of this Fe-oxo cluster stabilization.
This work was supported by the Spanish Government (RTI2018-096138-A-I00 and RTI2018-101049-B-I00) and the Regional Government of Madrid (TALENTO grants 2017-T1/IND-5148 and 2017-T1/AMB-5264) for funding. We acknowledge the computing facilities of CSUC for providing computational resources. XAS experiments were performed at the BL22 beamline (proposal 2019093796) at the ALBA Synchrotron with the collaboration of ALBA staff. We thank DESY and Dr M. Wharmby for assistance in using the beamline P02.1 for the PDF experiments (proposal I-20190239 EC). The research leading to this result has been supported by the project CALIPSOplus (Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020). C. C. B. acknowledges the European Social Funds and the Regional Government of Madrid for a postdoctoral contract (PEJD-2018-POST/IND-7909). I. R. M. acknowledges the Universidad Autónoma de Madrid for a predoctoral fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- F. A. Son, M. C. Wasson, T. Islamoglu, Z. Chen, X. Gong, S. L. Hanna, J. Lyu, X. Wang, K. B. Idrees, J. J. Mahle, G. W. Peterson and O. K. Farha, Chem. Mater., 2020, 32, 4609–4617 CrossRef.
- H. G. T. Ly, G. Fu, A. Kondinski, B. Bueken, D. D. Vos and T. N. Parac-Vogt, J. Am. Chem. Soc., 2018, 140, 6325–6335 CrossRef CAS.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- R. Gil-San-Millan, E. López-Maya, A. E. Platero-Prats, V. Torres-Pérez, P. Delgado, A. W. Augustyniak, M. K. Kim, H. W. Lee, S. G. Ryu and J. A. R. Navarro, J. Am. Chem. Soc., 2019, 141, 11801–11805 CrossRef CAS.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS.
- J. Jiang, F. Gándara, Y.-B. Zhang, K. Na, O. M. Yaghi and W. G. Klemperer, J. Am. Chem. Soc., 2014, 136(37), 12844–12847 CrossRef CAS.
- N. Van Velthoven, S. Waitschat, S. M. Chavan, P. Liu, S. Smolders, J. Vercammen, B. Bueken, S. Bals, K. P. Lillerud, N. Stock and D. E. De Vos, Chem. Sci., 2019, 10, 3616–3622 RSC.
- J. Liu, L. R. Redfern, Y. Liao, T. Islamoglu, A. Atilgan, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2019, 11, 47822–47829 CrossRef CAS.
- Z. Li, A. W. Peters, A. E. Platero-Prats, J. Liu, C. W. Kung, H. Noh, M. R. DeStefano, N. M. Schweitzer, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2017, 139, 15251–15258 CrossRef CAS.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176, 249–265 CrossRef.
- Y. Li, T. Yang, S. Qiu, W. Lin, J. Yan, S. Fan and Q. Zhou, Chem. Eng. J., 2020, 389(124382), 1–10 Search PubMed.
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Liu, J. T. Hupp, O. K. Farha, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 4178–4185 CrossRef CAS.
- H. Xu, S. Sommer, N. L. N. Broge, J. Gao and B. B. Iversen, Chem. – Eur. J., 2019, 25, 2051–2058 CrossRef CAS.
- C. Castillo-Blas, J. M. Moreno, I. Romero-Muñiz and A. E. Platero-Prats, Nanoscale, 2020, 12, 15577–15587 RSC.
- B. Jeon, Q. V. Overmeere, A. C. T. V. Duin and S. Ramanathan, Phys. Chem. Chem. Phys., 2013, 15, 1821–1830 RSC.
- K. M. Ø. Jensen, H. L. Andersen, C. Tyrsted, E. D. Bøjesen, A. C. Dippel, N. Lock, S. J. L. Billinge, B. B. Iversen and M. Christensen, ACS Nano, 2014, 8, 10704–10714 CrossRef CAS.
- H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS.
- A. E. Platero-Prats, A. B. League, V. Bernales, J. Ye, L. C. Gallington, A. Vjunov, N. M. Schweitzer, Z. Li, J. Zheng, B. L. Mehdi, A. J. Stevens, A. Dohnalkova, M. Balasubramanian, O. K. Farha, J. T. Hupp, N. D. Browning, J. L. Fulton, D. M. Camaioni, J. A. Lercher, D. G. Truhlar, L. Gagliardi, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2017, 139, 10410–10418 CrossRef CAS.
- J. K. Kowalska, A. W. Hahn, A. Albers, C. E. Schiewer, R. Bjornsson, F. A. Lima, F. Meyer and S. DeBeer, Inorg. Chem., 2016, 55, 4485–4497 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- R. N. Collins, K. M. Rosso, A. L. Rose, C. J. Glover and T. David Waite, Geochim. Cosmochim. Acta, 2016, 177, 150–169 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. Destefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06134d |
| This journal is © The Royal Society of Chemistry 2020 |