
Nickel-catalyzed insertions of vinylidenes into Si–H bonds†
Sourish
Biswas‡
 ,
Sudipta
Pal‡
and
Christopher
Uyeda
*
,
Sudipta
Pal‡
and
Christopher
Uyeda
*
Department of Chemistry, Purdue University, 560 Oval Dr., West Lafayette, IN, USA. E-mail: cuyeda@purdue.edu
First published on 27th October 2020
Abstract
A nickel-catalyzed reductive cyclization of 1,1-dichloroalkenyl silanes is reported. The products of this reaction are unsaturated five- or six-membered silacycles. Intermolecular variants are also described, providing access to trisubstituted vinyl silanes that are not accessible by alkyne hydrosilylation or sila-Heck-type processes. A variety of silanes can be utilized, including those that serve as nucleophilic partners in Hiyama cross-coupling reactions. Mechanistic studies using deuterium-labelled silanes are described.
Substitutions of carbon for silicon have attracted the interest of medicinal chemists as an avenue to optimize the potency and pharmacokinetic properties of lead compounds (Fig. 1A).1 The larger atomic radius of silicon causes subtle changes in geometry and conformation, impacting target binding affinity.2 Silicon substitution generally increases lipophilicity, which can improve cellular uptake with polar compounds.3 Finally, the incorporation of silicon into five- and six-membered rings blocks oxidative aromatization, a common metabolic liability.4 Despite these potential benefits, the effect of silicon substitution is often unpredictable, and there are currently no silicon-containing drugs and only a few silicon-containing agrochemicals5 that have successfully reached market. Future investigations of silicon in biologically active molecules will depend on the availability of robust synthetic methods that facilitate the construction of Si–C bonds.6
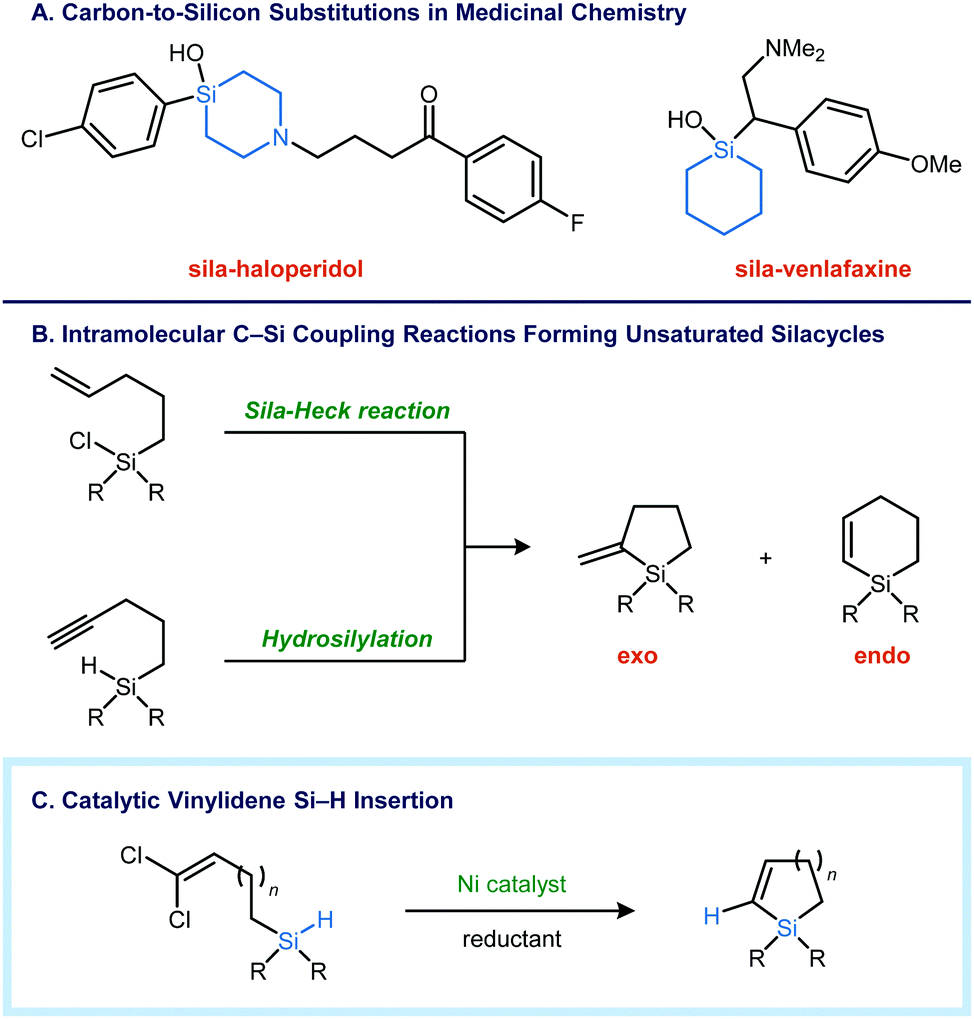 | ||
| Fig. 1 Nickel-catalyzed reductive insertions of 1,1-dichloroalkenes into Si–H bonds as a route to unsaturated silacycles. | ||
Unsaturated silicon heterocycles are commonly synthesized using intramolecular C–C bond forming reactions, where silicon is incorporated into the acyclic precursor as a tethering atom.7 Alternatively, direct intramolecular Si–C coupling can be carried out using an alkyne hydrosilylation8 or a sila-Heck type process (Fig. 1B).9 In both of these approaches, endo and exo cyclization modes are possible, and the selectivity is dependent on myriad factors, such as the tether length, the substitution pattern of the π-bond, steric effects, and the presence of functional groups that impart electronic bias.
Recently, we have showed that transition metal vinylidene complexes can be generated from 1,1-dichloroalkenes and a metal reductant such as Zn.10 We reasoned that the intramolecular insertion of such M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCR2 species into a Si–H bond11 would yield an unsaturated silacycle without the regioselectivity issues that arise with additions across alkenes and alkynes. To that end, we report here a nickel-catalyzed reductive cyclization of 1,1-dichloroalkenyl silanes to form five- and six-membered silacycles (Fig. 1C). The catalytic reductive Si–H insertion can also be carried out in an intermolecular context to provide trisubstituted vinyl silanes, which are not accessible by alkyne hydrosilylation.
CCR2 species into a Si–H bond11 would yield an unsaturated silacycle without the regioselectivity issues that arise with additions across alkenes and alkynes. To that end, we report here a nickel-catalyzed reductive cyclization of 1,1-dichloroalkenyl silanes to form five- and six-membered silacycles (Fig. 1C). The catalytic reductive Si–H insertion can also be carried out in an intermolecular context to provide trisubstituted vinyl silanes, which are not accessible by alkyne hydrosilylation.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditionsa | Yield (2) (%) |
| a Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), Ni(dme)Cl2 (0.05 equiv.), (±)-t-BuQuinox (0.06 equiv.), Zn (4.0 equiv.), DMA (0.2 mL), Et2O (0.6 mL), rt, 16 h. Yields were determined by 1H NMR integration against a mesitylene internal standard. | ||
| 1 | None | 91 |
| 2 | No Zn | 0 |
| 3 | No Ni(dme)Cl2 | 0 |
| 4 | No (±)-t-BuQuinox (3) | 0 |
| 5 | Mn instead of Zn | 85 |
| 6 | DMA only instead of Et2O/DMA | 36 |
| 7 | t-BuPyrox (4) instead of 3 | 50 |
| 8 | BnBiox (5) instead of 3 | 17 |
| 9 | BenzoQuinox (6) instead of 3 | 83 |
| 10 | DIPPIP (7) instead of 3 | 45 |
| 11 | PhPhen (8) instead of 3 | 43 |
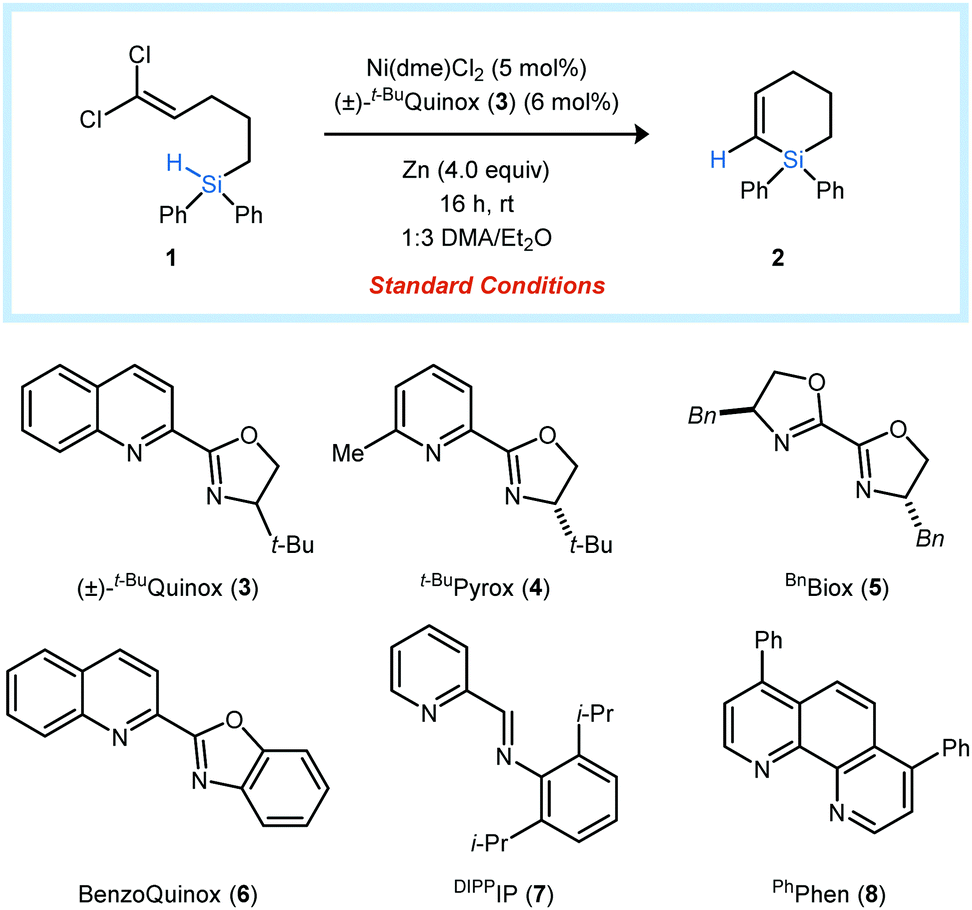
The dichloroalkenyl silane 1 was selected as a model substrate for the development of a catalytic reductive vinylidene Si–H insertion reaction. Under optimized conditions, (t-BuQuinox)NiCl2 (5 mol%) promotes the cyclization of 1 to form the six-membered unsaturated silacycle 2 in 91% yield (entry 1). Control experiments indicated that the reductant, metal source, and supporting ligand are required for the reaction to proceed. Mn is a viable reductant but is slightly less effective than Zn (entry 5). A combination of Et2O and a polar amide solvent, such as DMA, gave the highest yields, and the use of DMA alone resulted in a significant decrease in product formation (entry 6). t-BuQuinox (3) proved to be the optimal ligand. However, other bidentate nitrogen-donor ligands also promoted the formation of 2, with yields ranging from 17–83% (entries 7–11).
With optimized conditions in hand, we investigated the substrate scope of the reaction (Fig. 2). Five- and six-membered rings could be generated in high yield. Two examples of seven-membered ring-forming reactions were demonstrated, providing products 11 and 15 in modest yields of 29% and 40%, respectively. The substituents on Si could be alkyl, aryl, or a combination of the two.
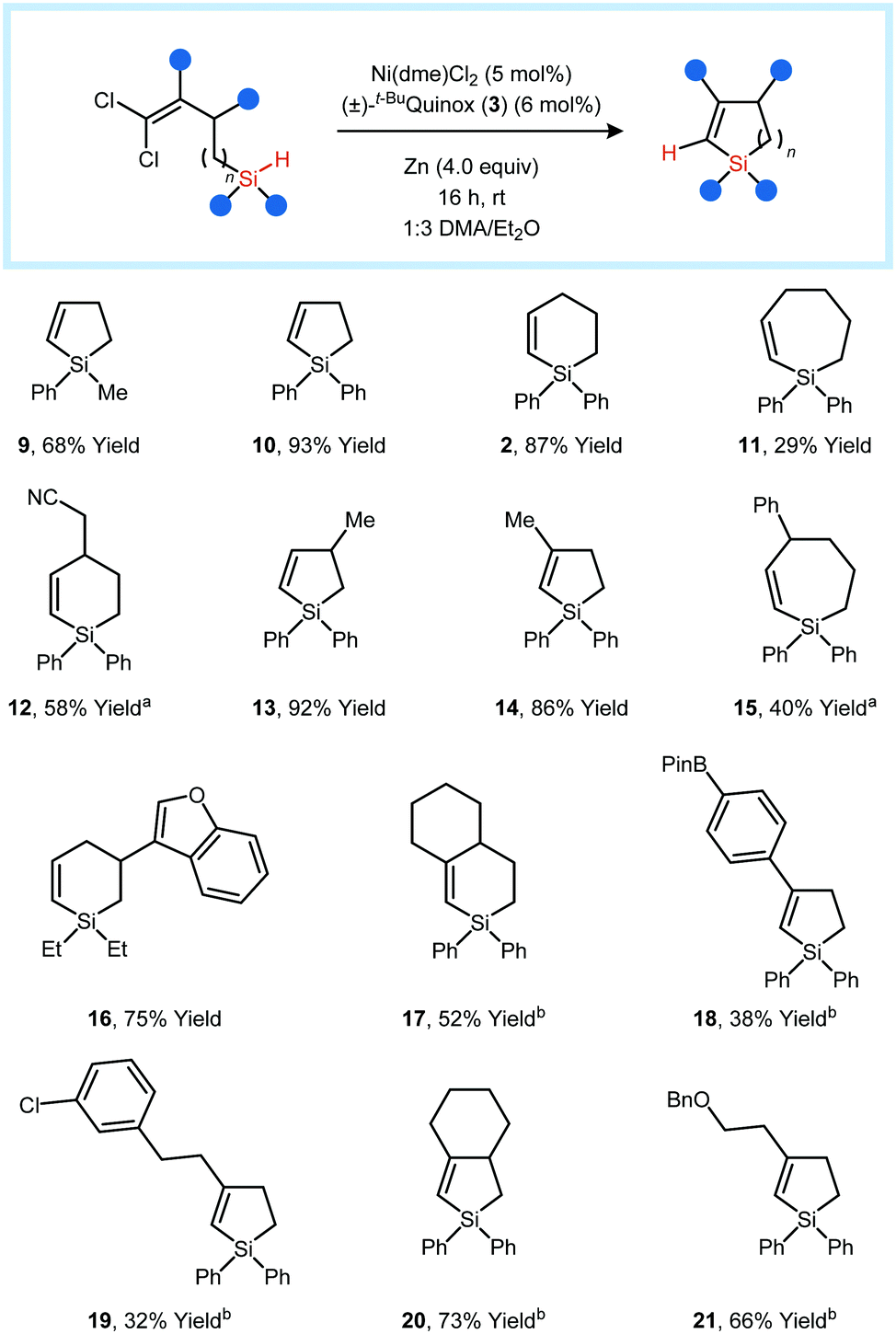 | ||
Fig. 2 Substrate scope studies. Reactions were conducted using the standard conditions shown in Table 1. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 mol% catalyst loading. b20 mol% catalyst loading; 50 °C temperature. 10 mol% catalyst loading. b20 mol% catalyst loading; 50 °C temperature. | ||
Intermolecular variants of the vinylidene Si–H insertion were also investigated (Fig. 3A). In order to obtain high yields, it was necessary to make minor modifications to the reaction conditions developed for the intramolecular reaction: the catalyst loading was increased from 5 mol% to 10 mol%, the co-solvent was changed from Et2O to THF, and the reaction temperature and concentration were increased. Under this set of conditions, vinylsilane 22 was obtained in 75% yield from a tropinone-derived 1,1-dichloroalkene and dimethylphenylsilane. Other tertiary silanes and other ketone-derived 1,1-dichloroalkenes also proved to be viable as reaction partners. Benzyldimethylsilane could be used as a substrate, and the Si–H insertion product can engage in Hiyama cross-coupling reactions (Fig. 3B).12
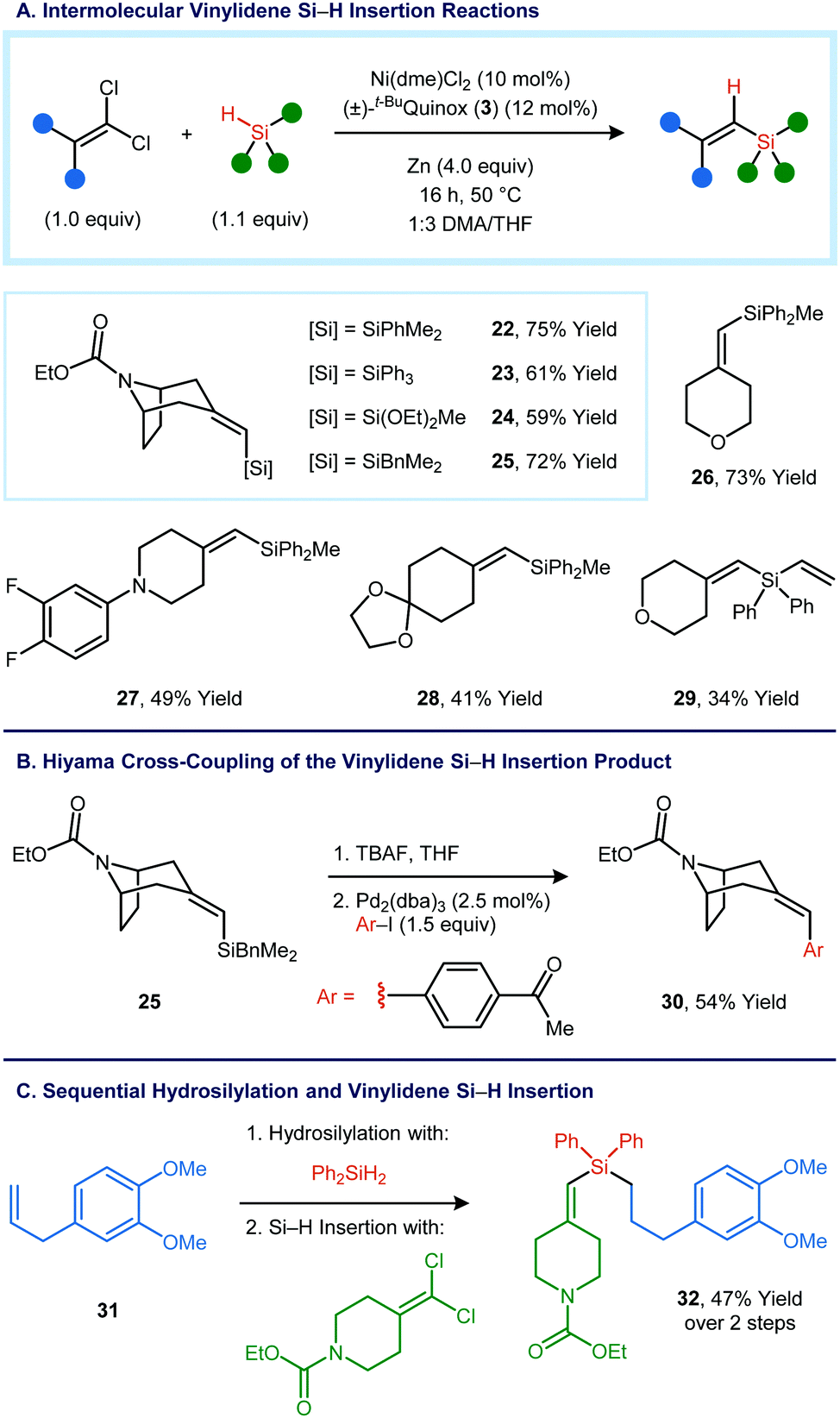 | ||
| Fig. 3 (A) Intermolecular vinylidene Si–H insertion reactions, providing trisubstituted vinylsilanes. (B) Synthesis of a vinylsilane that can be utilized as a nucleophilic partner in a Hiyama cross-coupling reaction. (C) Sequential hydrosilylation and vinylidene Si–H insertion reactions of a secondary organosilane. | ||
Sequential C–Si bond-forming reactions were carried out using a secondary organosilane. Catalytic hydrosilylation of methyl eugenol (32) using diphenylsilane yielded a primary organosilane,13 which could then be used in an intermolecular Si–H insertion reaction to form 32 in 47% yield over two steps (Fig. 3C).
Experiments using a deuterium labelled silane were carried out in order to gain insight into the mechanism of Si–H insertion.14 As expected, the reaction between 1,1-dichloroalkene 33 and Ph2MeSiD yielded product 26-d1 with >99% deuterium incorporation at the vinyl position (Fig. 4A). A kinetic isotope effect (KIE) competition experiment was carried out using a 1:1 mixture of Ph2MeSiH and Ph2MeSiD (Fig. 4B). The product (26) contained a 1:1 mixture of H and D at the vinyl position, indicating a kH/kD of 1.0. This result appears to be inconsistent with a concerted insertion of a metal vinylidene into the Si–H bond, regardless of whether the insertion is rate-determining or occurs in a fast step following rate-determining generation of the metal vinylidene.15,16
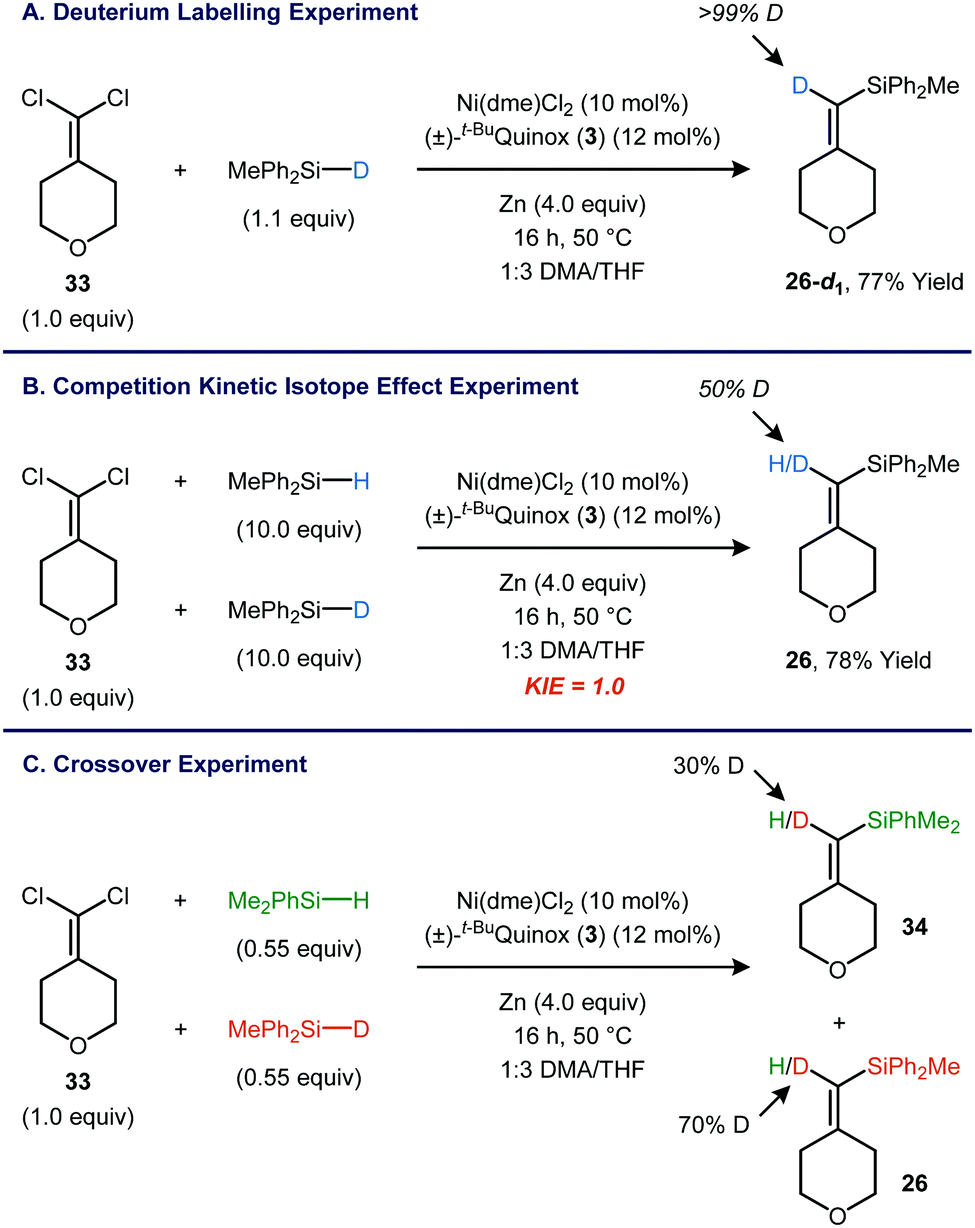 | ||
| Fig. 4 Mechanistic studies using a deuterium-labelled silane. | ||
Interestingly, scrambling was observed when a reaction was carried out using a 1:1 mixture of PhMe2SiH and Ph2MeSiD (Fig. 4C). The PhMe2Si-substituted product contains 30% D, and the Ph2MeSi-substituted product is correspondingly enriched in H. When the PhMe2SiH/Ph2MeSiD mixture was subjected to the standard catalytic conditions in the absence of the 1,1-dichloroalkene 33, H/D scrambling was also observed, indicating that the catalyst is able to activate the Si–H bond without having to generate a metal vinylidene. One scenario that would be consistent with both the KIE and scrambling experiments is a pre-equilibrium Si–H oxidative addition that occurs prior to the rate-limiting step. This rate-limiting step could involve activation of the 1,1-dichloroalkene or formation of the C–Si bond.
In summary, (Quinox)Ni catalysts promote reductive insertions of 1,1-dichloroalkenes into Si–H bonds. One synthetic application of this transformation is in the synthesis of unsaturated five- and six-membered silacycles. Additionally, moderate yields were obtained for the synthesis of seven-membered silacycles, which have not previously been prepared by other methods of ring-closure. Ongoing investigations are aimed at elucidating the mechanism of Si–H insertion and extending this reactivity to other bond insertion reactions.
This research was supported by the NIH (R35 GM124791). C. U. acknowledges support from a Camille Dreyfus Teacher-Scholar award and a Lilly Grantee award. We thank Annah Kalb and Celia He for helpful discussion and experimental assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS; (b) J. S. Mills and G. A. Showell, Expert Opin. Invest. Drugs, 2004, 13, 1149–1157 CrossRef CAS; (c) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS; (d) S. Gately and R. West, Drug Dev. Res., 2007, 68, 156–163 CrossRef CAS; (e) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS.
- (a) J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Organometallics, 2006, 25, 1188–1198 CrossRef CAS; (b) C. Zhou, J. Cheng, R. Beadle, F. G. Earley, Z. Li and P. Maienfisch, Bioorg. Med. Chem., 2020, 28, 115509 CrossRef CAS.
- S. J. Barraza and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 6668–6684 CrossRef CAS.
- T. Johansson, L. Weidolf, F. Popp, R. Tacke and U. Jurva, Drug Metab. Dispos., 2010, 38, 73–83 CrossRef CAS.
- (a) W. K. Moberg, G. S. Basarab, J. Cuomo and P. H. Liang, Synthesis and Chemistry of Agrochemicals, American Chemical Society, 1987, ch. 26, vol. 355, pp. 288–301 Search PubMed; (b) S. M. Sieburth, C. J. Manly and D. W. Gammon, Pestic. Sci., 1990, 28, 289–307 CrossRef CAS.
- (a) D. Liu and S. A. Kozmin, Angew. Chem., Int. Ed., 2001, 40, 4757–4759 CrossRef CAS; (b) H. Fang, W. Hou, G. Liu and Z. Huang, J. Am. Chem. Soc., 2017, 139, 11601–11609 CrossRef CAS; (c) M. Gimferrer, Y. Minami, Y. Noguchi, T. Hiyama and A. Poater, Organometallics, 2018, 37, 1456–1461 CrossRef CAS; (d) T. Ohmura, I. Sasaki and M. Suginome, Org. Lett., 2019, 21, 1649–1653 CrossRef CAS; (e) H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2019, 58, 4695–4699 CrossRef CAS; (f) I. Sasaki, T. Ohmura and M. Suginome, Org. Lett., 2020, 22, 2961–2966 CrossRef.
- (a) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC; (b) D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 983–1010 CAS.
- (a) T. Sudo, N. Asao and Y. Yamamoto, J. Org. Chem., 2000, 65, 8919–8923 CrossRef CAS; (b) J. A. Marshall and M. M. Yanik, Org. Lett., 2000, 2, 2173–2175 CrossRef CAS; (c) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726–12727 CrossRef CAS; (d) S. E. Denmark and W. Pan, Org. Lett., 2002, 4, 4163–4166 CrossRef CAS; (e) B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2003, 125, 30–31 CrossRef CAS.
- W. B. Reid, J. R. McAtee and D. A. Watson, Organometallics, 2019, 38, 3796–3803 CrossRef CAS.
- (a) S. Pal, Y.-Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686–11689 CrossRef CAS; (b) C. M. Farley, K. Sasakura, Y.-Y. Zhou, V. V. Kanale and C. Uyeda, J. Am. Chem. Soc., 2020, 142, 4598–4603 CrossRef CAS.
- (a) M. S. Newman and C. D. Beard, J. Am. Chem. Soc., 1970, 92, 4309–4312 CrossRef CAS; (b) M. S. Newman and T. B. Patrick, J. Am. Chem. Soc., 1970, 92, 4312–4315 CrossRef CAS; (c) C. D. Beard and J. C. Craig, J. Am. Chem. Soc., 1974, 96, 7950–7954 CrossRef CAS; (d) P. J. Stang and A. E. Learned, J. Am. Chem. Soc., 1987, 109, 5019–5020 CrossRef CAS.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 53, 6104–6110 CrossRef.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS.
- (a) Y. Landais, L. Parra-Rapado, D. Planchenault and V. Weber, Tetrahedron Lett., 1997, 38, 229–232 CrossRef CAS; (b) L. A. Dakin, P. C. Ong, J. S. Panek, R. J. Staples and P. Stavropoulos, Organometallics, 2000, 19, 2896–2908 CrossRef CAS; (c) H. Keipour and T. Ollevier, Org. Lett., 2017, 19, 5736–5739 CrossRef CAS.
- J. C. Gilbert and D. H. Giamalva, J. Org. Chem., 1985, 50, 2586–2587 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and spectra. See DOI: 10.1039/d0cc05970f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |