
Boronic acid-mediated ring-opening and Ni-catalyzed arylation of 1-arylcyclopropyl tosylates†
L. Reginald
Mills
 ,
John J.
Monteith
and
Sophie A. L.
Rousseaux
*
,
John J.
Monteith
and
Sophie A. L.
Rousseaux
*
Davenport Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada. E-mail: sophie.rousseaux@utoronto.ca
First published on 30th September 2020
Abstract
Herein, we describe a protocol for the ring-opening arylation of 1-arylcyclopropyl tosylates, in which boronic acids promote ring-opening and a Ni catalyst facilitates arylation in high regioselectivity. A number of 2-arylated allyl derivatives are synthesized, which are relevant motifs found in biologically active molecules.
Cyclopropanols are well established synthetic intermediates that serve as three-carbon units for accessing a number of valuable products.1 A unique cyclopropanol-derived reagent is the cyclopropyl tosylate (Fig. 1a), which is readily accessed from an ester using Kulinkovich chemistry (retrosynthesis arrow), or from the corresponding ketone using Simmons–Smith chemistry.1 These intermediates are labile at the C–O bond, and can be activated with a Brønsted or Lewis acid in the presence of a nucleophile (Nu) to give the ring-opened, allyl product. An early example of this type of ring-opening functionalization is the solvolysis of cyclopropyl tosylates with acetic acid reported by DePuy and coworkers in 1965, which yields the allyl acetate.2,3 Since then, there have been a number of reports employing cyclopropyl tosylates,1,4,5 including studies of the mechanism of ring-opening,6 and metal-catalyzed substitution reactions.7 A different use of the ring-opening reactivity of cyclopropyl tosylates was reported by Kulinkovich and coworkers, who discovered that these intermediates can be converted to the 2-substituted allyl bromide in the presence of MgBr2.8 Other Lewis acids like MgCl2, TiCl4, and AlCl3 can also achieve ring-opening to generate the corresponding allyl halide.8 More recently, Cha and coworkers found that 1-alkenylcyclopropyl tosylates are amenable to ring-opening functionalization with nucleophiles such as amines in the presence of catalytic Pd.9 However, the scope of this transformation is limited to 1-alkenyl-substituted cyclopropyl tosylates.
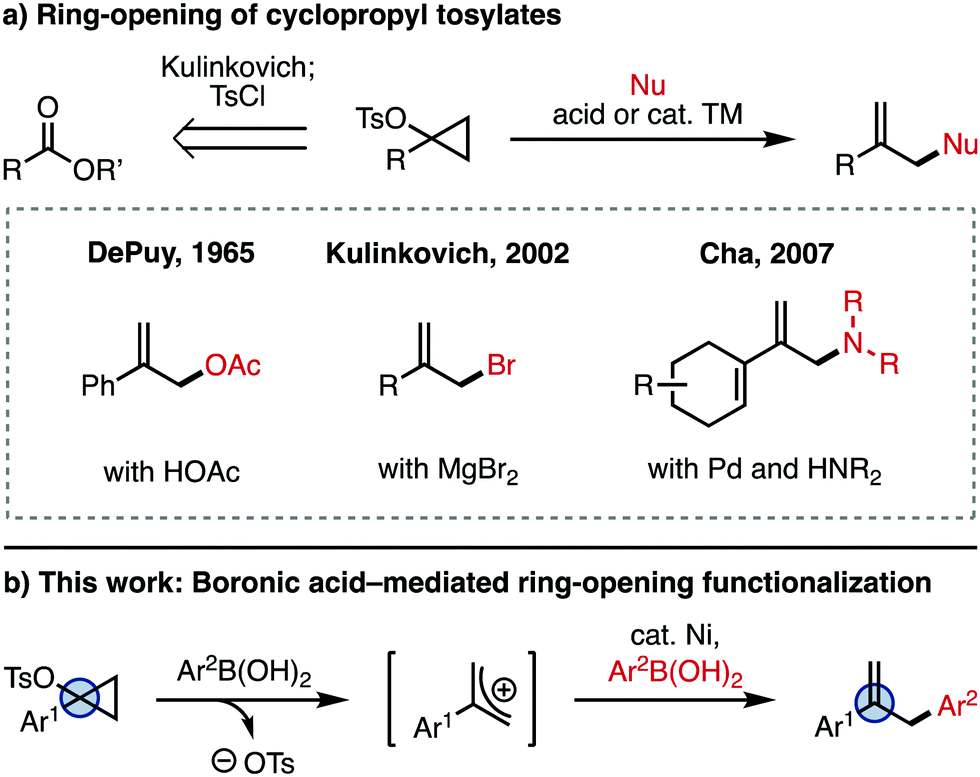 | ||
| Fig. 1 The 2-substituted allyl motif and synthetic approaches for its preparation. | ||
Herein, we report the ring-opening functionalization of 1-arylcyclopropyl tosylates. This process was discovered while we were exploring related chemistry for the use of cyclopropanols as C–O electrophiles in a Ni-catalyzed Negishi cross-coupling.10 For this reaction, the ring-opening is mediated by arylboronic acid (Ar2B(OH)2), which generates the allyl cation (Fig. 1b). The allyl cation then undergoes Ni-catalyzed cross-coupling with the arylboronic acid, giving the arylated product.11 Together, this reactivity is a simple and straightforward way of accessing allyl derivatives bearing a variety of aryl substituents at the 2-position, which are valuable synthetic intermediates.12,13 While other methods for the metal-catalyzed arylation of 2-aryl-substituted allyl electrophiles exist,14 rapid access to allyl coupling partners is not always trivial, since they are often synthesized in multiple steps from esters, ketones, or other oxidized derivatives. Since cyclopropyl tosylates are directly synthesized from esters or ketones,1 our strategy offers complementary access to 2-substituted allyl products.
Since some 1-substituted cyclopropyl tosylates can be ring-opened and functionalized using catalytic Pd,9 we began our investigation by exploring Pd catalysis to facilitate this reaction (Table 1). Using 1-phenylcyclopropyl tosylate (1a), 4-methoxyphenylboronic acid (2 equiv.), PdCl2(PPh3)2 (10 mol%), and K3PO4 (4 equiv.), the arylated product (2a) was obtained in a modest 22% yield (entry 1). Switching the catalyst to NiCl2(PPh3)2 delivered the product in excellent 96% yield (entry 2). Other Ni catalysts like NiCl2(PCy3)2 were also competent in this reaction, though performed slightly less efficiently (entry 3) (for other catalysts, see the ESI†). Intriguingly, the reaction was still efficient in the absence of catalyst, giving the product in 92% yield, though with imperfect selectivity for substitution at the ipso position (entry 4). Arylboronic acids are known to participate in SEAr chemistry with imperfect regioselectivity,15 suggesting that a similar process may be occurring under these catalyst-free conditions. Under catalyst-free conditions, we found that the aryl boroxine (entry 5) was less efficient than the arylboronic acid, affording product 2a in 33% yield. Arylboronic esters such as ArBnep (entry 6), as well as potassium aryl trifluoroborate salts (entry 7), were ineffective in this transformation. It should also be noted that cyclopropyl tosylate (1a) is stable at 110 °C after 1 h, both in the absence and presence of a Ni catalyst. In the presence of arylboronic acid at 110 °C, full conversion of this substrate is achieved after 1 h.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | [B] | Yield 2ab (%) |
para![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ortho :ortho |
| a Reactions performed on 0.10 mmol scale. See ESI for full details. b GC-MS yield using n-dodecane as internal standard. c Isolated yield. d 0.67 equiv. nep = neopentylglycol. | ||||
| 1 | PdCl2(PPh3)2 | B(OH)2 | 22 | — |
| 2 | NiCl2(PPh3)2 | B(OH)2 | 96 (82)c | >20:1 |
| 3 | NiCl2(PCy3)2 | B(OH)2 | 82 | >20:1 |
| 4 | None | B(OH)2 | 92 | 8.9:1 |
| 5 | None | (ArBO)3d | 33 | 7.6:1 |
| 6 | None | Bnep | <5 | — |
| 7 | None | BF3K | <5 | — |
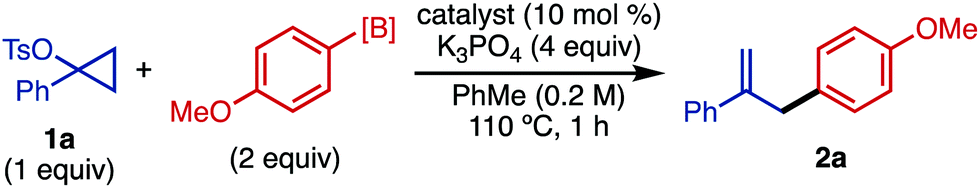
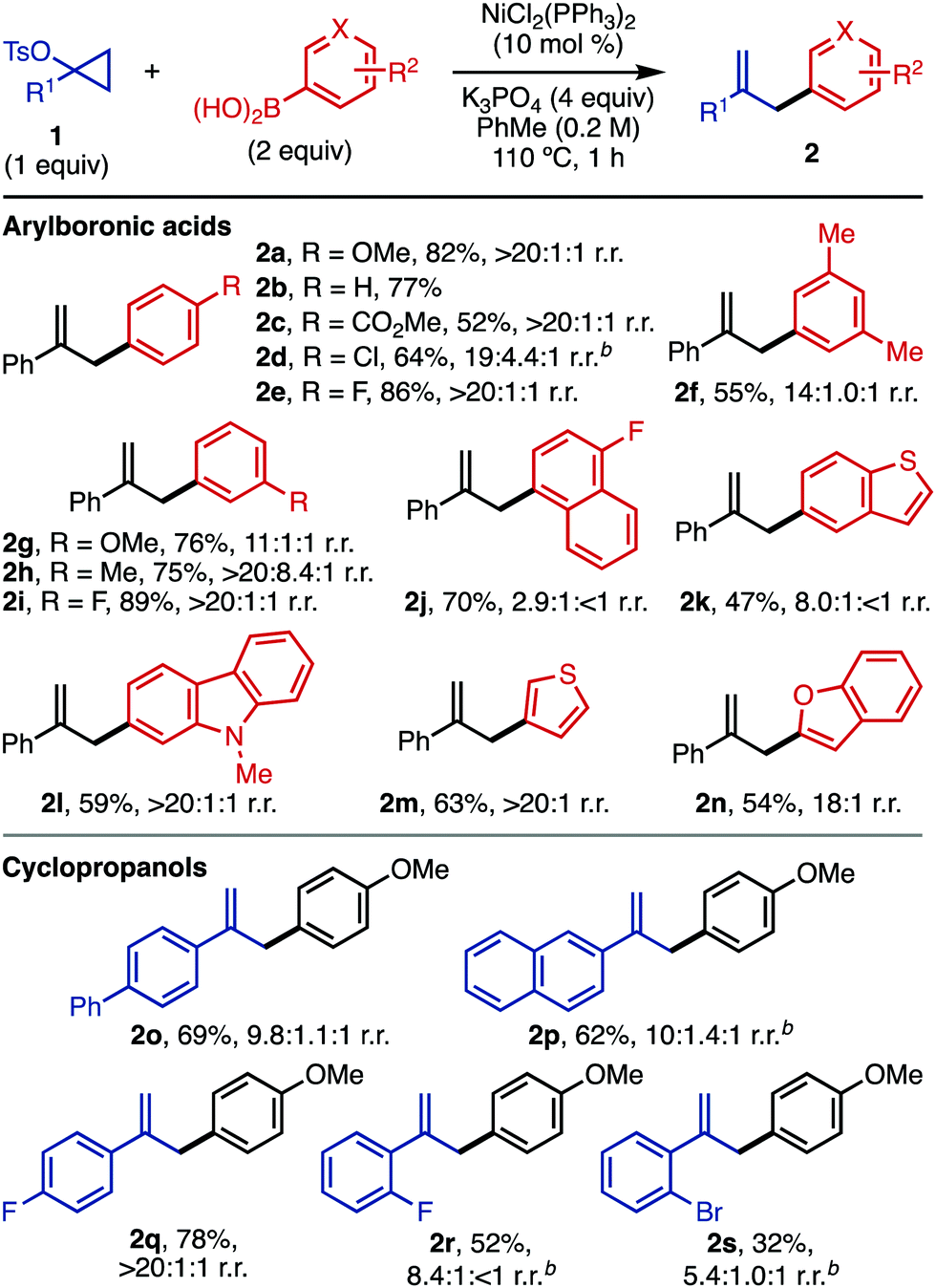
With these optimized conditions, we next explored the scope of this reaction (Table 2). Since some arylation is possible in the absence of a Ni catalyst (Table 1, entry 4), several entries gave mixtures of regioisomers. However, reactions in the presence of Ni are generally more regioselective than those carried out without catalyst. For the structures drawn, the substitution reflects the regioisomer obtained after ipso substitution of the boronic acid. The conditions are compatible with a number of arylboronic acids, including electron-rich (2a), electron-neutral (2b, 2f), and electron-deficient (2c–2e, 2j) substrates. Arylboronic acids with strong para electron-withdrawing groups (4-acetylphenyl, 4-cyanophenyl) gave <20% yield of the desired product. A number of meta-substituted arylboronic acids were also viable (2f–2i). For substrates with labile halogens like chlorides (2d), the reaction performed better in the absence of a Ni catalyst; in the presence of Ni, products resulting from halide functionalization (reduction, arylation) were detected by GC-MS. The compatibility of electron-withdrawing groups (2c) is complementary to related Friedel–Crafts chemistry of activated cyclopropanols using electron-rich arenes.11
| a Reactions performed on 0.20–0.40 mmol scale. Yields are isolated and the combination of regioisomers. Regioisomer ratios were determined by GC-MS analysis of the crude reaction mixture. The structures drawn reflect the regioisomer obtained after ipso substitution of the arylboronic acid. b Reaction performed without NiCl2(PPh3)2; for yields with and without Ni, see ESI. |
|---|
|
For this Ni-catalyzed process, the regioselectivity with respect to arylboronic acid functionalization with both electron-donating and -withdrawing groups is dictated by the position of the boronic acid and not the inherent reactivity expected for SEAr chemistry (e.g. ortho and para for 2f–2i; meta for 2c). Employing electron-deficient substrates (e.g.2c) in the absence of catalytic Ni gave low yields (<10%). A number of electron-rich heteroaromatic boronic acids are also compatible with the chemistry (2k–2n). For heteroaromatic boronic acids with rings that could participate in Friedel–Crafts chemistry (e.g., 2k, 2l), substitution was only detected on the boronic acid-containing ring.
A number of 1-arylcyclopropyl tosylates are compatible with the chemistry, including electron-neutral (2o, 2p) and electron-deficient (2q–2s) substrates.16ortho-Substituted 1-arylcyclopropyl tosylates (2r, 2s) afforded the desired product in higher yield in the absence of Ni catalyst (for yields and selectivity in the presence of catalytic Ni, see the ESI†). Cyclopropyl tosylates without 1-aryl substituents gave very little conversion under standard conditions, both in the presence and absence of NiCl2(PPh3)2. The low yields for some substrates may be due to decomposition of the cyclopropyl tosylate (1) at the reaction temperature (110 °C).
Next, mechanistic experiments were performed to better understand (i) the mechanism of the background Ni-free arylation reaction and (ii) the role of the boronic acid in this background process (Fig. 2). First, a head-to-head comparison was made for the reaction of 3-methoxyphenylboronic acid with and without a Ni catalyst (Fig. 2a). In the presence of 10 mol% NiCl2(PPh3)2 (entry 1), product 2g was obtained in 91% GC-MS yield and 11:1:1 selectivity for functionalization at the ipso position. The predominant formation of this regioisomer is consistent with transmetallation of 3-methoxyphenylboronic acid to Ni. Alternatively, in the absence of Ni (entry 2), the product was obtained in only 41% GC-MS yield, with the major regioisomers instead being those resulting from substitution para and ortho to the electron-donating methoxy substituent. This result is consistent with an SEAr mechanism for the Ni-free background process. It also suggests that the arylboronic acid is likely responsible for cyclopropane ring-opening to generate the allyl cation intermediate.
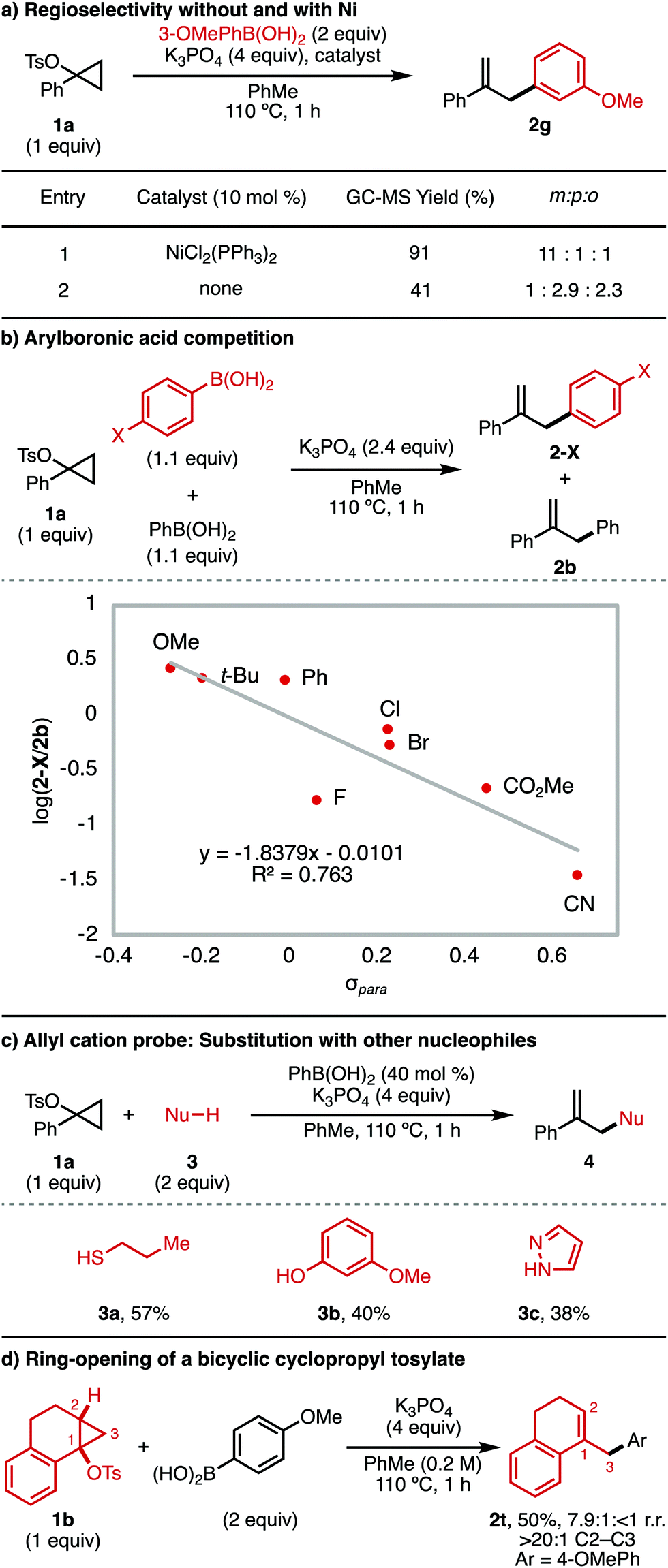 | ||
| Fig. 2 Mechanistic experiments. | ||
To better understand the Ni-free background reaction, competition experiments were performed between 4-substituted arylboronic acids (1.1 equiv.) and phenylboronic acid (1.1 equiv.). The product distribution between the 4-substituted aryl product (2-X) and the phenyl product (2b) was evaluated (Fig. 2b). When the log(2-X/2b) was taken and plotted according to σpara of 2-X, a negative linear relationship was obtained, consistent with build-up of positive charge in the product-determining step. This finding is consistent with an SEAr mechanism to yield the arylated product in the absence of Ni catalyst. It should be noted that in this experiment any electronic effects which promote boronic acid–mediated ring-opening of 1a cannot be excluded.
To further confirm the role of the arylboronic acid in ring-opening of the cyclopropyl tosylate, we investigated whether other nucleophiles could be used to trap an allyl cation generated from 1a using PhB(OH)2 (Fig. 2c). In the presence of substoichiometric amounts of phenylboronic acid (40 mol%) and K3PO4, 1-phenylcyclopropyl tosylate 1a underwent trapping with a range of nucleophiles (3) to yield the allyl product, 4. Viable nucleophiles include thiol (3a), phenol (3b),17 and pyrazole (3c).18
Finally, we examined whether the arylboronic acid-mediated cyclopropyl tosylate ring-opening was consistent with known regio- and stereoselectivity for acid-catalyzed ring-openings of cyclopropanol derivatives. Thus, a bicyclic cyclopropyl tosylate (1b) was exposed to the reaction conditions in the absence of Ni (Fig. 2d). The product obtained (2t) was that resulting from C2–C3 cleavage, with retention of the cis relationship between the 1-position and 2-position substituents. This result is consistent with conventional selectivity for SN1 reactions of cyclopropyl electrophiles, which are known to undergo concerted, disrotatory C2–C3 ring-opening to generate the allyl cation.5,6,19
A proposed mechanistic picture for this cyclopropyl tosylate ring-opening arylation is outlined in Fig. 3. Starting with 1-arylcyclopropyl tosylates (1), activation of the substrate occurs with arylboronic acid, which likely acts as an H-bond donor (5).20 This intermediate then fragments to release tosylate anion and allyl cation 6. In the absence of Ni, this cation can be trapped with an activated arylboronate to yield Wheland intermediate 7. Elimination of the boronate restores aromaticity, forming arylated product 2. Alternatively, in the presence of Ni(0), oxidative addition can yield Ni(II) intermediate 8, which can undergo transmetallation with arylboronic acid to yield intermediate 9. Finally, reductive elimination releases the allyl arene 2. In the presence of Ni, it is likely that some of the non-Ni-catalyzed process also occurs, which explains the imperfect regioselectivity for certain entries in Table 2.
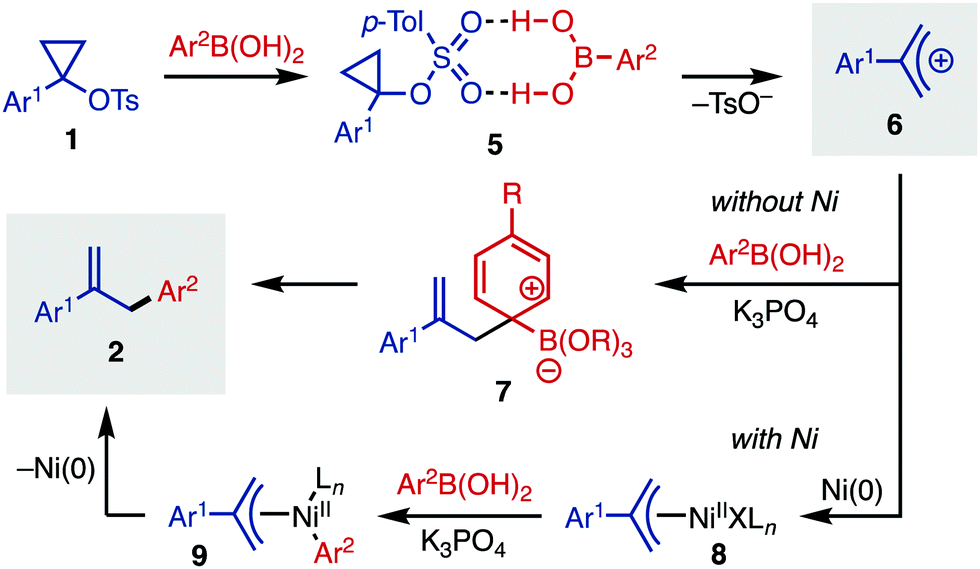 | ||
| Fig. 3 Proposed mechanisms without and with Ni. | ||
In conclusion, we have discovered an arylboronic acid-mediated and Ni-catalyzed process for the ring-opening arylation of 1-arylcyclopropyl tosylates, which are readily accessible from esters via the Kulinkovich reaction. The arylboronic acid plays two roles in this reaction: (i) it enables cyclopropane ring-opening and (ii), it participates in a Ni-catalyzed cross-coupling to yield allyl products bearing aryl substituents at the 2 position.
We thank NSERC (Discovery Grants and Canada Research Chair programs), the Canada Foundation for Innovation (Project No. 35261), the Ontario Research Fund, Kennarshore Inc., and the University of Toronto for generous financial support of this work. We also acknowledge the Canada Foundation for Innovation (Project No. 19119) and the Ontario Research Fund for funding the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers. L. R. M. thanks NSERC for a graduate scholarship (PGS-D). J. J. M. thanks NSERC for a graduate scholarship (CGS M) and the University of Toronto for a doctoral scholarship (FAST).
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- (a) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef CAS; (b) A. Nikolaev and A. Orellana, Synthesis, 2016, 1741–1768 CAS; (c) L. R. Mills and S. A. L. Rousseaux, Eur. J. Org. Chem., 2019, 8 CrossRef CAS.
- C. H. DePuy, L. G. Schnack, J. W. Hausser and W. Wiedemann, J. Am. Chem. Soc., 1965, 87, 4006 CrossRef CAS.
- For the ring-opening of cyclopropyl tosylate: J. D. Roberts and V. C. Chambers, J. Am. Chem. Soc., 1951, 73, 5034 CrossRef CAS.
- (a) B. A. Howell and J. G. Jewett, J. Am. Chem. Soc., 1971, 93, 798 CrossRef; (b) J. Salaün, J. Org. Chem., 1976, 41, 1237 CrossRef.
- D. H. Gibson and C. H. DePuy, Chem. Rev., 1974, 74, 605 CrossRef CAS.
- (a) J. W. Hausser and N. J. Pinkowski, J. Am. Chem. Soc., 1967, 89, 6981 CrossRef CAS; (b) P. v. R. Schleyer, W. F. Sliwinski, G. W. Van Dine, U. Schöllkopf, J. Paust and K. Fellenberger, J. Am. Chem. Soc., 1972, 94, 125 CrossRef CAS.
- (a) A. Stolle, J. Salaün and A. de Meijere, Tetrahedron Lett., 1990, 31, 4593 CrossRef CAS; (b) P. Aufranc, J. Ollivier, A. Stolle, C. Bremer, M. Es-Sayed, A. de Meijere and J. Salaün, Tetrahedron Lett., 1993, 34, 4193 CrossRef CAS; (c) S. Racouchot, J. Ollivier and J. Salaün, Synlett, 2000, 1729 CAS; (d) F. Lecornué, F. Charnay-Pouget and J. Ollivier, Synlett, 2006, 1407 Search PubMed.
- (a) Y. Y. Kozyrkov and O. G. Kulinkovich, Synlett, 2002, 443 CrossRef CAS; (b) O. G. Kulinkovich, Y. Y. Kozyrkov, A. V. Bekish, E. A. Matiushenkov and I. L. Lysenko, Synthesis, 2004, 1713 Search PubMed.
- L. G. Quan, H. G. Lee and J. K. Cha, Org. Lett., 2007, 9, 4439 CrossRef CAS.
- L. R. Mills, J. J. Monteith, G. D. Passos Gomes, A. Aspuru-Guzik and S. A. L. Rousseaux, J. Am. Chem. Soc., 2020, 142, 13246 CrossRef CAS.
- For a related SEAr functionalization reaction of silyl-protected cyclopropanols using electron-rich arenes and diethylaminosulfur trifluoride (DAST) to generate the allyl cation, see M. Kirihara, T. Noguchi, H. Kakuda, T. Akimoto, A. Shimajiri, M. Morishita, A. Hatano and Y. Hirai, Tetrahedron Lett., 2006, 47, 3777–3780 CrossRef CAS.
- A. A. Protter and S. Chakravarty, Compounds and Methods of Treating Hypertension. WO Patent 2012112961, 2012 Search PubMed.
- C. S. Burgey, D. V. Paone, A. Q. Shaw, J. Z. Deng, D. N. Nguyen, C. M. Potteiger, S. L. Graham, J. P. Vacca and T. M. Williams, Org. Lett., 2008, 10, 3235 CrossRef CAS.
- For select recent metal-catalyzed reactions employing 2-substituted allyl electrophiles: (a) C. Dong, L. Zhang, X. Xue, H. Li, Z. Yu, W. Tang and L. Xu, RSC Adv., 2014, 4, 11152 RSC; (b) X. Cui, S. Wang, Y. Zhang, W. Deng, Q. Qian and H. Gong, Org. Biomol. Chem., 2013, 11, 3094 RSC; (c) M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211 CrossRef CAS; (d) X.-G. Jia, P. Guo, J. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640 RSC.
- G. Wu, S. Xu, Y. Deng, C. Wu, X. Zhao, W. Ji, Y. Zhang and J. Wang, Tetrahedron, 2016, 72, 8022–8030 CrossRef CAS.
- In our hands, electron-rich 1-(hetero)arylcyclopropyl tosylates (e.g., 1-(4-methoxyphenyl), 1-(2-thienyl), 1-(2-furanyl)) were difficult to access.
- The reaction with phenol 3b gave the O-allylated product.
- Adding catalytic NiCl2(PPh3)2 to these reactions gave no noticeable increase in yield. Nucleophilic arenes that typically participate efficiently in Friedel–Crafts chemistry (e.g. 1H-indole, 1,3,5-trimethoxybenzene) yielded no detectable amount of substituted product.
- I. V. Alabugin, Stereoelectronic Effects. Chichester, UK, Hoboken, NJ, John Wiley & Sons, 2016 Search PubMed.
- (a) K. M. Diemoz and A. K. Franz, J. Org. Chem., 2019, 84, 1126 CrossRef CAS; (b) S. Zhang, D. Lebœuf and J. Moran, Chem. – Eur. J., 2020, 26, 9883 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05895e |
| This journal is © The Royal Society of Chemistry 2020 |