
Ascorbic acid-mediated reductive disassembly of Fe3+-tannic acid shells in degradable single-cell nanoencapsulation†
Hojae
Lee
,
Joohyouck
Park
,
Sang Yeong
Han
,
Sol
Han
,
Wongu
Youn
,
Hyunwoo
Choi
,
Gyeongwon
Yun
and
Insung S.
Choi
 *
*
Center for Cell-Encapsulation Research, Department of Chemistry, KAIST, Daejeon 34141, Korea. E-mail: ischoi@kaist.ac.kr
First published on 22nd September 2020
Abstract
Rapid degradation of Fe3+-tannic acid films is achieved under mild conditions via ascorbic acid-mediated Fe3+ reduction, which overcomes the problems in the disassembly of a metal–organic complex including slow reaction rates and reaction incompatibility with living cells. The strategy of reductive disassembly is applied to degradable single-cell nanoencapsulation, providing an advanced tool for tightly controlling and manipulating the cell–material interface.
Single-cell nanoencapsulation (SCNE) aims to protect living cells by chemically constructing ultrathin (<100 nm), durable artificial shells at the single-cell level and also to confer on them exogenous (bio)chemical functions that are not innate in the cells.1,2 Artificial exoskeletons have recently evolved to degrade in response to external stimuli, after temporal cytoprotection of single cells, mimicking the germination process of dormant spores found in nature. In degradable SCNE, supramolecular coordination polymers, such as metal–organic frameworks (MOFs) and metal-phenolic complexes (MPCs), have intensively been used because of the reversible nature of metal–ligand coordination bonds.3–8 Falcaro et al. encapsulated Saccharomyces cerevisiae and Micrococcus luteus within a MOF shell of zeolitic imidazolate framework (ZIF-8), composed of Zn2+ and 2-methylimidazole, which was degraded by the addition of ethylenediaminetetraacetic acid (EDTA).3,4 EDTA was also employed by Brinker et al. for the degradation of MOF nanoparticle shells, which were cross-linked by tannic acid (TA), on HeLa and other mammalian cells.5 We and others reported the use of the Fe3+-TA MPC for degradable SCNE, where the MPC shell was disassembled by EDTA or HCl.6–8 Collectively, these approaches could be categorized as a ligand-modification strategy, which exchanges the ligands (e.g., substitution of TA or 2-methylimidazole with EDTA) or decreases the ligand affinity (e.g., protonation of catecholate to catechol in TA by HCl or its oxidation to quinone).9,10
On the other hand, nature has developed sophisticated processes for control over iron (Fe) species.11,12 For example, certain bacteria (e.g., Escherichia coli and Salmonella typhimurium) shuttle Fe species between their cytoplasm and the extracellular region by precisely controlling the coordination complex formation of Fe3+ and the siderophores, named enterobactins, which are composed of three catecholate ligands with 2,6,10-trioxo-1,5,9-trioxacyclododecane as a core.13 The active transport of Fe3+ into the cytosol is performed by the strong Fe3+-enterobactin complex (Kf ≈ 52), structurally reminiscent of the Fe3+-TA MPC.14 Mechanisms of Fe release in the cytosol are still not resolved completely, but studies indicate the involvement of Fe3+ reduction, leading to the formation of a loosely bound Fe2+ complex and subsequent Fe2+ release.15,16 Inspired by this biological mechanism, in this work, we suggest an alternative metal-modification strategy to the cytocompatible degradation of Fe3+-TA MPC shells in SCNE. Specifically, we demonstrate that Fe3+ reduction to Fe2+ in the Fe3+-TA MPC by vitamin C (L-ascorbic acid, AA) disassembles the MPC rapidly under physiologically relevant conditions, and apply the Fe3+ reduction-based method to in situ shell degradation on S. cerevisiae.
We first investigated the reduction characteristics of the Fe3+-TA MPC upon addition of AA to the MPC solution. The solution, initially dark purple, turned colorless within 3 h, as observed by the naked eye, indicating the disassembly of the Fe3+-TA MPC (Fig. 1a). The ligand-to-metal charge-transfer (LMCT) band at 565 nm of the Fe3+-TA MPC sharply decreased in intensity after addition of AA (Fig. 1b). The results indicated that the reduction of Fe3+ to Fe2+ in the MPC decreased the charge density of the Fe metal center, changing the hard to borderline hard/soft Lewis acid, and leading to a decreased affinity for the negatively charged, hard oxygen donor in TA. The lower reactivity of Fe2+ to TA and the lower stability of the Fe2+-TA complex enabled the dissociation of the MPC. The rapidity of MPC disassembly was confirmed by the ellipsometric-thickness measurement (Fig. 1c). The Fe3+-TA MPC films, on a flat gold substrate, prepared using our iron gall ink (IGI) strategy that uses Fe2+ as an Fe source,17–19 were incubated in an aqueous AA solution (concentration: 1, 5, or 10 mM), and the film thickness was measured at a predetermined time. The ellipsometric analysis arguably showed that the film degraded rapidly, upon AA addition, in a concentration-dependent manner. For example, complete film degradation was observed within 4 min with 10 mM AA, while 30% of the film remained even after 1 h of incubation with 1 mM AA. The comparative studies showed that the degradation efficiency of 1 mM AA was comparable to that of EDTA (10 mM) or gly-HCl buffer (pH 2), indicating the far better performance of the metal-modification strategy over the ligand-modification one. The film degradation was additionally confirmed by field-emission scanning electron microscopy (FE-SEM) and contact angle goniometry (Fig. S1, ESI†). Our method was also applied seamlessly to other Fe3+-TA MPC structures, such as hollow capsules and free-standing films: only 19% of the capsules remained after 12 h of incubation in the 10 mM AA solution, but 65% and 86% remained in the EDTA solution (10 mM) and the gly-HCl buffer (pH 2), respectively (Fig. S2, ESI†). The 2.5 μm-thick free-standing film degraded completely within 2 h in the AA solution, while 46% and 65% of the film still remained in the EDTA solution and gly-HCl buffer, respectively (Fig. S3, ESI†). We thought that the partially cleaved MPC species remained present at the oil–water interface for stabilization of the system and led to the observed slow degradation for hollow capsules. The film degradation was additionally demonstrated by the successful fabrication of Fe3+-TA MPC micropatterns through AA-mediated wet etching for 1 min (Fig. S4, ESI†).
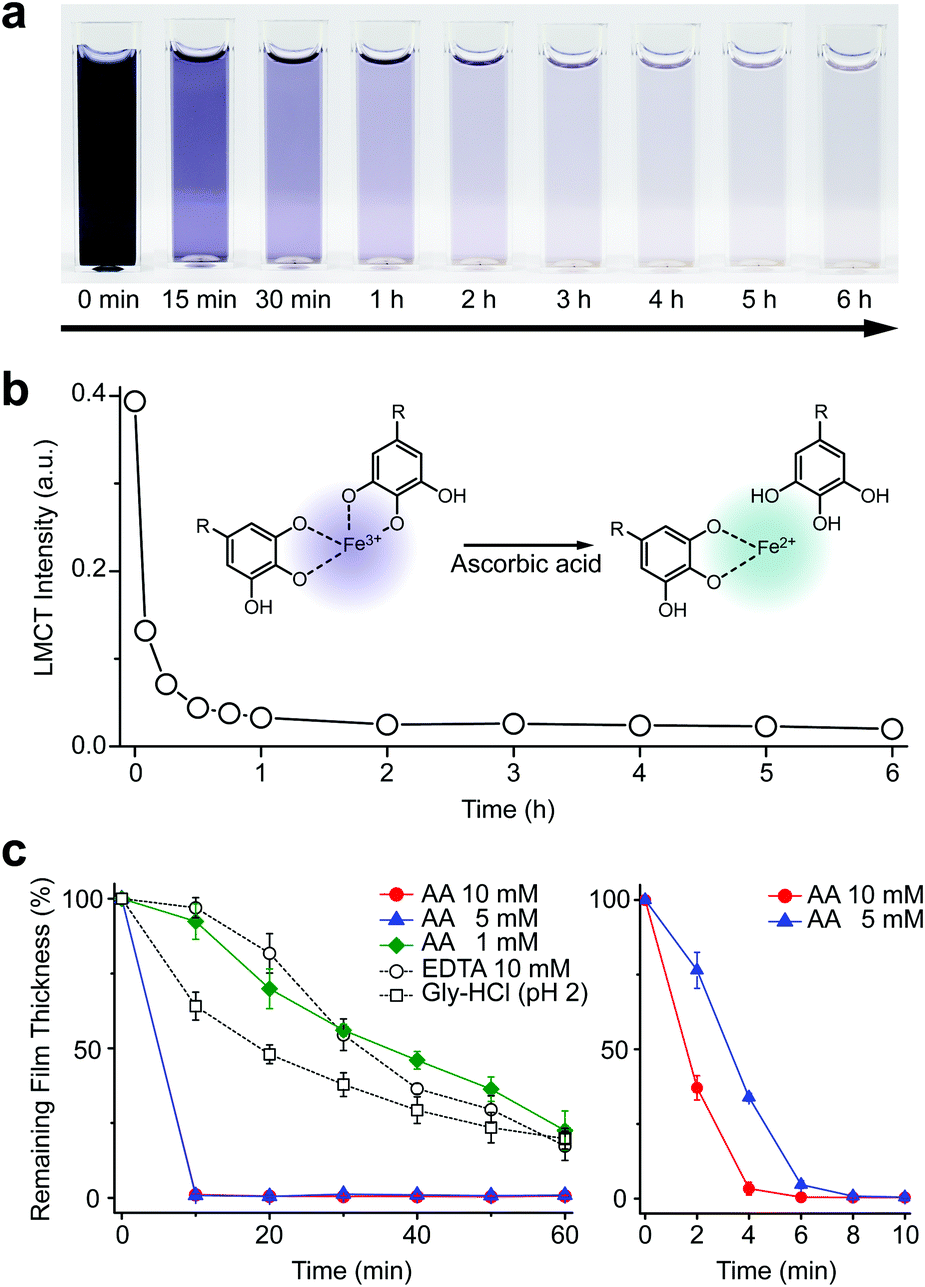 | ||
| Fig. 1 (a) Time-lapse optical images of the Fe3+-TA MPC solution after addition of ascorbic acid at 0 min (10 mM). (b) Graph of LMCT intensity at 565 nm versus reaction time. (c) Graphs of film thickness versus reaction time. The Fe3+-TA MPC film was incubated in an aqueous solution of AA (1, 5, or 10 mM), EDTA (10 mM), or gly-HCl buffer (pH 2). The right graph for AA (5 and 10 mM) is the enlarged one for the first 10 min of incubation. | ||
We tested other biological reductants, such as NADH and glutathione (GSH), for MPC degradation in vain (Fig. S5, ESI†). In biological systems, oxidoreductase and FAD are required for NADH to reduce the Fe3+ complex.20 It is reported that GSH works for Fe3+ reduction only in an oxygen-free atmosphere,21 while ascorbate prefers to maintain the Fe2+-ascorbate complex, not the Fe3+ complex, in the presence of oxygen.22 Our failed results, along with the previous reports, supported the suggested active role of AA in Fe metabolism, in which other biological reductants usually act to regenerate AA from dehydroascorbate (an oxidized form of AA), rather than being involved directly in Fe3+ reduction.23,24 The redox cycle of AA was demonstrated in the Fe3+-TA film degradation by the redox coupling between AA and GSH in the solution (Fig. S6, ESI†). The addition of GSH (10 mM) to the AA solution (0.1 mM) led to noticeably increased film degradation. The pH dependency of our system additionally supported the direct involvement of AA in Fe3+ reduction and MPC disassembly. It has been reported that the reduction potential of Fe3+ in the Fe3+-catecholate complex is −790 mV at pH 7.4, and it increases as the pH decreases (e.g., −570 mV at pH 6 and 170 mV at pH 4).25 Accordingly, AA with a reduction potential of 90 mV was observed to be operational in the film degradation at pH values less than 5 (Fig. S5, ESI†).24
The AA-mediated, reductive disassembly of the Fe3+-TA MPC was directly applied to SCNE, providing a cytocompatible strategy for shell degradation (Fig. 2a). The Fe3+-TA MPC shell was formed on S. cerevisiae (leading to the construction of yeast@Fe3+-TA), confirmed by FE-SEM and confocal laser-scanning microscopy (CLSM). For the SCNE of S. cerevisiae, the direct formation of the Fe3+-TA complex with Fe3+ was used instead of the IGI strategy, the coating rate of which is relatively low.6 The CLSM images, after treatment with bovine serum albumin (BSA)-Alexa Fluor 647 (red) for shell visualization and fluorescein diacetate (FDA, green) for viability assay, clearly showed that individual S. cerevisiae cells were encapsulated uniformly by the Fe3+-TA MPC without any noticeable decrease in cell viability, in accordance with our previous report (Fig. 2b, left). The AA addition to the yeast@Fe3+-TA suspension changed the suspension color from purple to pale white, indicating the disassembly of the Fe3+-TA MPC and its shell degradation (Fig. S7, ESI†). The CLSM image showed the complete disappearance of the ring-shaped red fluorescence, confirming the cytocompatible shell degradation (Fig. 2b, right). As expected from the green fluorescence, the cell viability did not decrease after shell degradation (Fig. S8, ESI†). The FE-SEM images further confirmed that the nanoparticulate morphology of the Fe3+-TA MPC shell disappeared completely after AA treatment (Fig. 2c).
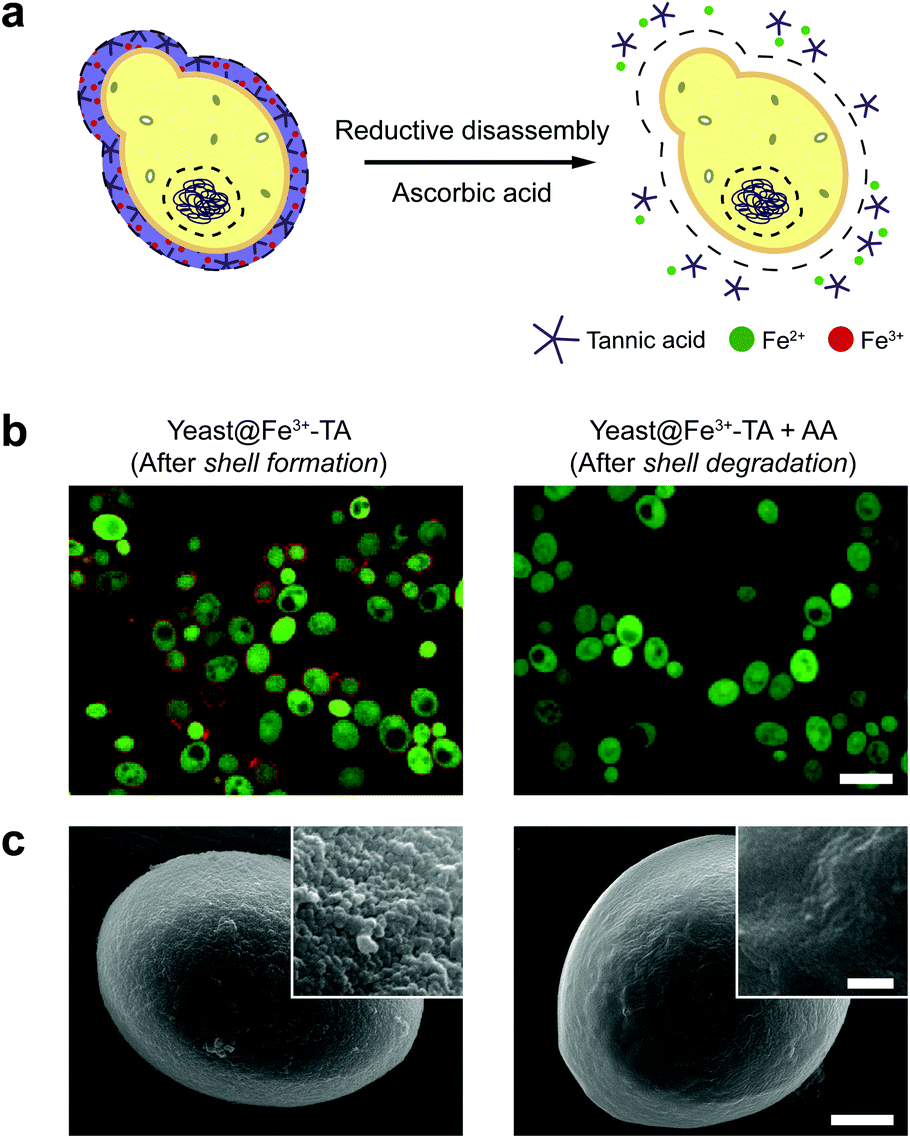 | ||
| Fig. 2 (a) Schematic representation of the reduction-based Fe3+-TA shell degradation on S. cerevisiae with ascorbic acid. (b) CLSM and (c) FE-SEM images of S. cerevisiae after shell formation and degradation. In (b), the Fe3+-TA shell was stained with BSA-Alexa Fluor 647 (red) for shell visualization and S. cerevisiae was stained with FDA (green) for viability assay. Scale bar: 10 μm. In (c), scale bar: 1 μm, and 100 nm for the inset. | ||
It should be noted that the previous methods including ours, using potentially toxic chemicals (i.e., HCl and EDTA) for shell degradation, required totally separated processes of shell degradation and cell culture steps, hampering their use in in vivo and practical applications. For example, yeast@Fe3+-TA was first immersed for 3 h in HCl solution (pH 2) to remove the MPC shell and, after several washing steps, then incubated in a yeast-extract-peptone-dextrose (YPD) broth for culture.6 This was also true for EDTA.3 Our control experiments clearly showed that the direct addition of HCl or EDTA to the yeast@Fe3+-TA-suspended YPD broth (YPD_MPC[+]) restrained cell growth and proliferation (Fig. 3a). The microbial growth was monitored by optical density measurements at 600 nm (OD600). The OD600 values of YPD_MPC[+] did not change even after 30 h of incubation when EDTA or HCl was added (solid triangles in Fig. 3a). We thought that EDTA and HCl would be lethal to the yeast cells that started to bud and were budding, which was supported by the result that no growth was observed with native yeast under the same conditions (YPD_MPC[−], open triangles). In stark contrast, AA addition to YPD_MPC[+] enabled both shell degradation and cell proliferation concurrently (solid circle), without any negative effects on cell growth (open circle). All three samples (i.e., native yeast, native yeast with AA, and yeast@Fe3+-TA with AA) had a similar, if not the same, lag phase of 12 h. The retardation and restoration of cell growth by shell formation and degradation, which are desired in the artificial spores of SCNE,26,27 were investigated by calculating the  value that is defined as the time when ln(OD600) reaches −2.0 (Fig. 3b).28 The shell formation increased the
value that is defined as the time when ln(OD600) reaches −2.0 (Fig. 3b).28 The shell formation increased the 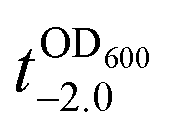 value to 20.5 h, and it returned back to the natural
value to 20.5 h, and it returned back to the natural 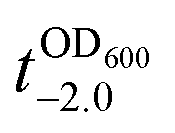 value after the addition of AA to YPD_MPC[+]. The growth rate (μ) was unperturbed (0.60 h−1 for native yeast; 0.56 h−1 after shell formation; 0.54 h−1 after shell degradation), additionally supporting the fact that the reductive disassembly process did not alter the metabolic activities. Taken together, the nature-inspired, AA-mediated reductive strategy provided a one-pot, cytocompatible tool for disassembling Fe3+-TA MPC films and shells.
value after the addition of AA to YPD_MPC[+]. The growth rate (μ) was unperturbed (0.60 h−1 for native yeast; 0.56 h−1 after shell formation; 0.54 h−1 after shell degradation), additionally supporting the fact that the reductive disassembly process did not alter the metabolic activities. Taken together, the nature-inspired, AA-mediated reductive strategy provided a one-pot, cytocompatible tool for disassembling Fe3+-TA MPC films and shells.
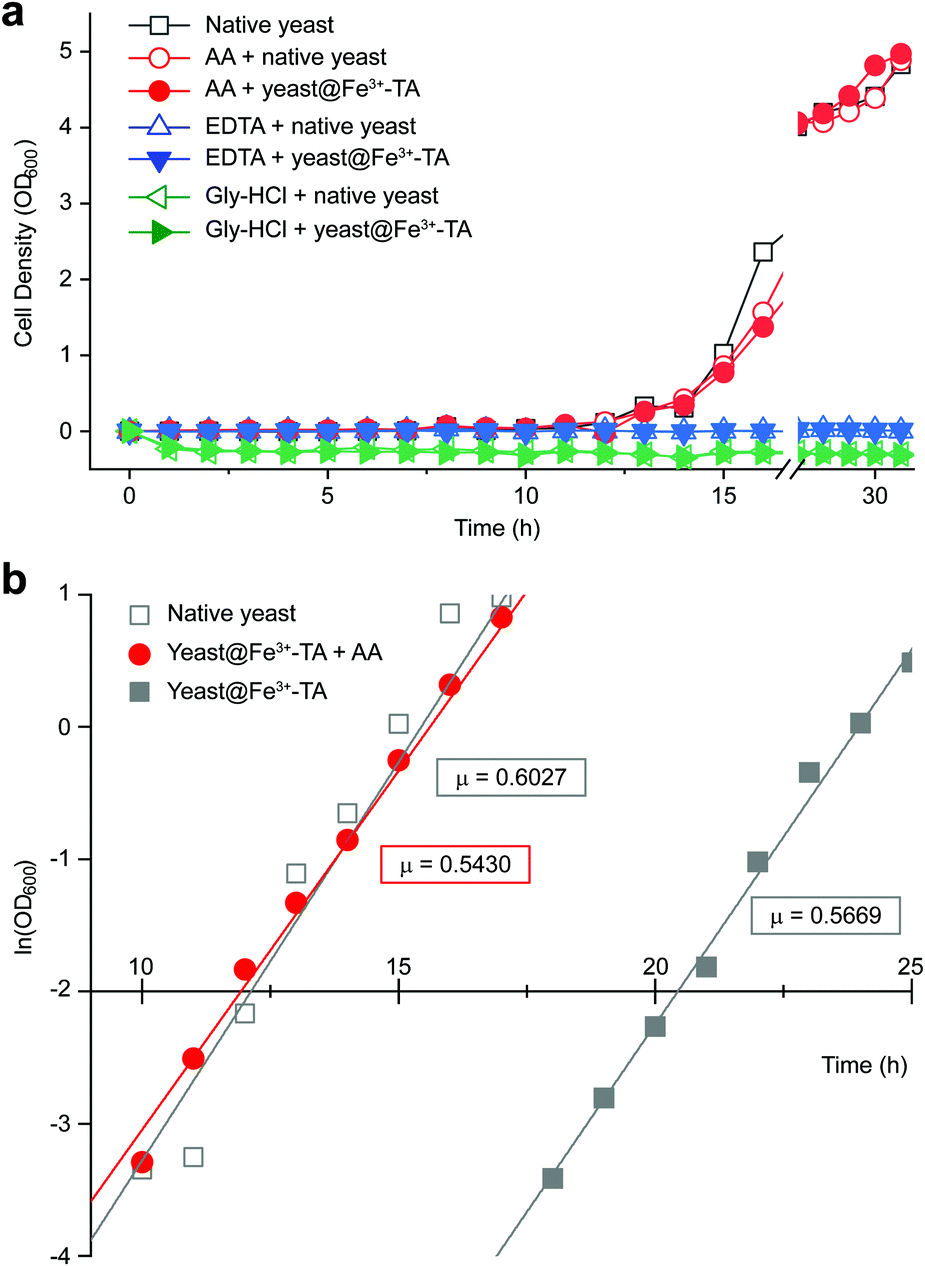 | ||
| Fig. 3 (a) Growth curve of yeast@Fe3+-TA in the YPD broth (YPD_MPC[+]) with AA (100 mM, solid red circle), EDTA (100 mM, solid blue triangle), or gly-HCl (pH 2, solid green triangle). The data for native yeast incubated in the YPD broth (YPD_MPC[−]) with AA, EDTA, or gly-HCl are depicted as open symbols for each case and those for native yeast in the as-prepared YPD broth are depicted as open grey squares. (b) Linear-fitted plots from −4.0 to 1.0 of ln(OD600) of native yeast (open grey square), and yeast@Fe3+-TA incubated in the as-prepared YPD (solid grey square) or the AA-added YPD broth (solid red circle). The slope, μ, is expressed in ln units per hour, corresponding to the cell-growth rate in the exponential growth phase. | ||
The reductive disassembly of the Fe3+-TA MPC could be coupled reversibly with its oxidative assembly (Fig. 4). We formed the Fe3+-TA MPC using the IGI strategy,17 in which Fe2+ as a precursor of Fe3+ was air-oxidized to the Fe3+ cation that formed a strong coordination complex with TA. The AA-mediated Fe3+ reduction enabled the film degradation (or the retardation of film growth, Fig. S9, ESI†) only until the complete consumption of AA, and the film growth was restored. The results confirmed that AA reacted only with Fe3+ and the process was redox-based.
 | ||
| Fig. 4 Graphs of film thickness versus reaction time in the IGI-based solution ([Fe2+] = 10 mM, [TA] = 1 mM). AA (0.1, 0.5, or 1 mM) was added to the solution after 3 h of reaction. | ||
In summary, we suggested a metal-modification strategy for the controlled disassembly of the Fe3+-TA MPC, inspired by the process of biological iron uptake in nature. The specific Fe3+ reduction to Fe2+ by a biological reductant, L-ascorbic acid (AA), disassembled the MPC rapidly under physiologically relevant conditions. The AA-mediated reductive disassembly solves the issues in the use of MPC-based hierarchical structures, especially in bio-related practical applications, exemplified herein by a cytocompatible, one-step, in situ MPC shell degradation in SCNE. The scope of our method is expandable to other metal–ligand coordination structures, such as MOFs (Fig. S10, ESI†) and supramolecular hydrogels.29–32 We, therefore, believe that its simplicity and wide applicability promise new vistas in the field of materials science,33–35 not to mention providing an advanced tool for chemical manipulation of living cells.
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP2012R1A3A2026403).
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Youn, J. Y. Kim, J. Park, N. Kim, H. Choi, H. Cho and I. S. Choi, Adv. Mater., 2020, 32, 1907001 CrossRef CAS.
- B. J. Kim, H. Cho, J. H. Park, J. F. Mano and I. S. Choi, Adv. Mater., 2018, 30, 1706063 CrossRef.
- K. Liang, J. J. Richardson, J. Cui, F. Caruso, C. J. Doonan and P. Falcaro, Adv. Mater., 2016, 28, 7910 CrossRef CAS.
- K. Liang, J. J. Richardson, C. J. Doonan, X. Mulet, Y. Ju, J. Cui, F. Caruso and P. Falcaro, Angew. Chem., Int. Ed., 2017, 129, 8630 CrossRef.
- W. Zhu, J. Guo, S. Amini, Y. Ju, J. O. Agola, A. Zimpel, J. Shang, A. Noureddine, F. Caruso, S. Wuttke, J. G. Croissant and C. J. Brinker, Adv. Mater., 2019, 31, 1900545 CrossRef.
- J. H. Park, K. Kim, J. Lee, J. Y. Choi, D. Hong, S. H. Yang, F. Caruso and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 12420 CAS.
- J. Lee, H. Cho, J. Choi, D. Hong, D. Kim, J. H. Park, S. H. Yang and I. S. Choi, Nanoscale, 2015, 7, 18918 RSC.
- W. Li, W. Bing, S. Huang, J. Ren and X. Qu, Adv. Funct. Mater., 2015, 25, 3775 CrossRef CAS.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154 CrossRef CAS.
- P. V. Cherepanov, Md. A. Rahim, N. Bertleff-Zieschang, Md. A. Sayeed, A. P. O’Mullane, S. E. Moulton and F. Caruso, ACS Appl. Mater. Interfaces, 2018, 10, 5828 CrossRef CAS.
- K. N. Raymond, B. E. Allred and A. K. Sia, Acc. Chem. Res., 2015, 48, 2496 CrossRef CAS.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413 CrossRef CAS.
- R. J. Abergel, J. A. Warner, D. K. Shuh and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 8920 CrossRef CAS.
- W. R. Harris, C. J. Carrano, S. R. Cooper, S. R. Sofen, A. E. Avdeef, J. V. McArdle and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 6097 CrossRef CAS.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584 CrossRef CAS.
- D. J. Ecker, B. F. Matzanke and K. N. Raymond, J. Bacteriol., 1986, 167, 666 CrossRef CAS.
- H. Lee, W. I. Kim, W. Youn, T. Park, S. Lee, T.-S. Kim, J. F. Mano and I. S. Choi, Adv. Mater., 2018, 30, 1805091 CrossRef.
- B. J. Kim, J. K. Lee and I. S. Choi, Chem. Commun., 2019, 55, 2142 RSC.
- S. Y. Han, S.-P. Hong, E. K. Kang, B. J. Kim, H. Lee, W. I. Kim and I. S. Choi, Cosmetics, 2019, 6, 23 CrossRef CAS.
- M. J. Kobylarz, G. A. Heieis, S. A. Loutet and M. E. P. Murphy, ACS Chem. Biol., 2017, 12, 1778 CrossRef CAS.
- M. Y. Hamed, R. C. Hider and J. Silver, Inorg. Chim. Acta, 1982, 66, 13 CrossRef CAS.
- J. E. Gorman and F. M. Clydesdale, J. Food Sci., 1983, 48, 1217 CrossRef CAS.
- C. H. Foyer and G. Noctor, Plant Physiol., 2011, 155, 2 CrossRef CAS.
- G. Noctor, Plant, Cell Environ., 2006, 29, 409 CrossRef CAS.
- C.-W. Lee, D. J. Ecker and K. N. Raymond, J. Am. Chem. Soc., 1985, 107, 6920 CrossRef CAS.
- J. H. Park, D. Hong, J. Lee and I. S. Choi, Acc. Chem. Res., 2016, 49, 792 CrossRef CAS.
- S. H. Yang, D. Hong, J. Lee, E. H. Ko and I. S. Choi, Small, 2013, 9, 178 CrossRef CAS.
- J. Lee, S. H. Yang, S.-P. Hong, D. Hong, H. Lee, H.-Y. Lee, Y.-G. Kim and I. S. Choi, Macromol. Rapid Commun., 2013, 34, 1351 CrossRef CAS.
- W. Zhu, J. Guo, J. O. Agola, J. G. Croissant, Z. Wang, J. Shang, E. Coker, B. Motevalli, A. Zimpel, S. Wuttke and C. J. Brinker, J. Am. Chem. Soc., 2019, 141, 7789 CrossRef CAS.
- W. Zhu, G. Xiang, J. Shang, J. Guo, B. Motevalli, P. Durfee, J. O. Agola, E. N. Coker and C. J. Brinker, Adv. Funct. Mater., 2018, 28, 1705274 CrossRef.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651 CrossRef CAS.
- A. Anderson, M. Krogsgaard and H. Birkedal, Biomacromolecules, 2018, 19, 1402 CrossRef.
- J. Guo, T. Suma, J. J. Richardson and H. Ejima, ACS Biomater. Sci. Eng., 2019, 5, 5578 CrossRef CAS.
- Md. A. Rahim, S. L. Kristufek, S. Pan, J. J. Richardson and F. Caruso, Angew. Chem., Int. Ed., 2019, 58, 1904 CrossRef CAS.
- Md. A. Rahim, G. Lin, P. P. Tomanin, Y. Ju, A. Barlow, M. Björnmalm and F. Caruso, ACS Appl. Mater. Interfaces, 2020, 12, 3746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary figures. See DOI: 10.1039/d0cc05856d |
| This journal is © The Royal Society of Chemistry 2020 |