
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arylation of aryllithiums with S-arylphenothiazinium ions for biaryl synthesis†
Tatsuya
Morofuji
 *a,
Tatsuki
Yoshida
a,
Ryosuke
Tsutsumi
b,
Masahiro
Yamanaka
b and
Naokazu
Kano
*a
*a,
Tatsuki
Yoshida
a,
Ryosuke
Tsutsumi
b,
Masahiro
Yamanaka
b and
Naokazu
Kano
*a
aDepartment of Chemistry, Faculty of Science, Gakushuin University, 1-5-1 Mejiro, Toshima-ku, Tokyo 171-8588, Japan. E-mail: tatsuya.morofuji@gakushuin.ac.jp; naokazu.kano@gakushuin.ac.jp
bDepartment of Chemistry, Faculty of Science, Rikkyo University, 3-34-1 Nishi-Ikebukuro, Toshima-ku, Tokyo 171-8501, Japan
First published on 13th October 2020
Abstract
Aryllithiums are one of the most common and important aryl nucleophiles; nevertheless, methods for arylation of aryllithums to produce biaryls have been limited. Herein, we report arylation of aryllithiums with S-arylphenothiazinium ions through selective ligand coupling of intermediary sulfuranes. Various unsymmetrical biaryls could be obtained without transition-metal catalysis.
Aryllithiums are one of the most common and important aryl nucleophiles in organic chemistry. Aryllithiums are known to react with various electrophiles, such as alkyl halides, carbonyl compounds, chlorosilanes and trimethoxyborane.1 However, methods for arylation of aryllithums to produce biaryls are very limited. Arylation of aryllithiums based on nucleophilic aromatic substitution2 or nucleophilic attack to aryne3 was reported, but there were few options regarding the applicable substrates. Although a few palladium-catalyzed methods have been reported,4 development of electrophilic arylation reagents that react with aryllithiums in the absence of transition metals is a simple but challenging problem (Fig. 1a).5
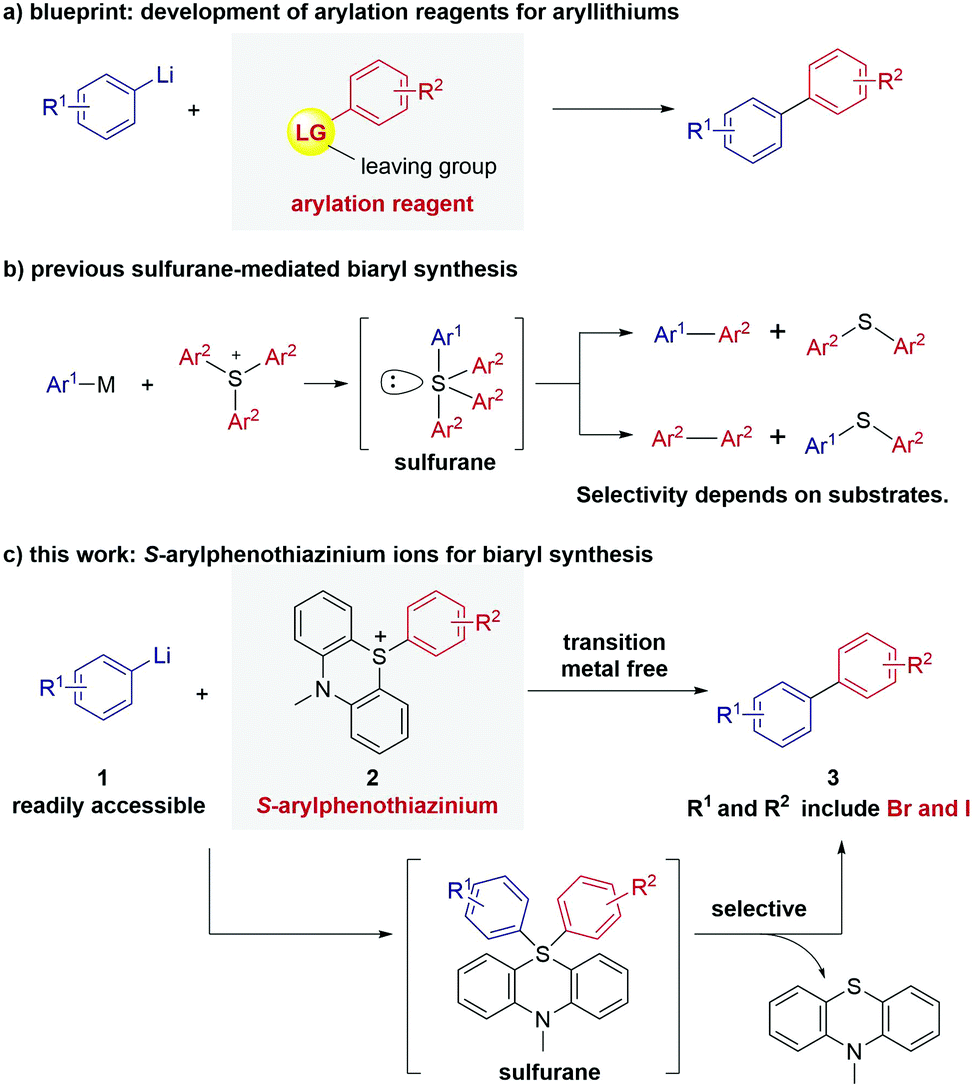 | ||
| Fig. 1 Arylation of aryllithiums. (a) Our blueprint: development of arylation reagents for aryllithiums. (b) Previous work: reactivity of sulfurane and its limitation for the synthesis of biaryl. (c) This work: S-arylphenothiazinium ions used as electrophilic arylation reagents for synthesizing unsymmetrical biaryls. | ||
In sulfur chemistry, the ligand coupling of tetraarylsulfuranes (IUPAC nomenclature: tetraaryl-λ4-sulfanes) has been reported.6 Reaction of triarylsulfoniums with arylmetal reagents including aryllithiums affords the tetraarylsulfurane intermediate, which decomposes to the corresponding biaryls and diaryl sulfides without transition-metal catalysis (Fig. 1b).7 However, these processes have very limited applications because the selectivity of the resulting products depends on aromatic substituents on the sulfur atom.7b,f One of the reasons why control of the selectivity of the ligand coupling is difficult is that conformation of sulfurane is not fixed through Berry's pseudorotation.7a To obtain unsymmetrical biaryls, previous ligand coupling was limited to use electron-poor heteroaromatic rings such as pyridine, azole, or quinoline as coupling partner, which promote desired ligand coupling.7i,8 In contrast, the selective synthesis of unsymmetrical biphenyl derivatives from sulfonium salts and aryl metals is a long-standing problem.
Herein, we report that S-arylphenothiazinium ions can solve the problem and be used as versatile arylation reagents for aryllithiums. The reaction of aryllithiums 1 with S-arylphenothiazinium ions 2 selectively afforded the corresponding biaryls 3 and phenothiazine (Fig. 1c). In contrast to the transition-metal-catalyzed approach, bromo- and iodo-substituted biaryls, which are attractive synthetic intermediates, could be synthesized using the present method. A computational study was performed to reveal the reaction mechanism of this sulfurane-mediated biaryl-formation.
First, we examined the reaction of some cyclic sulfonium ions9 with phenyllithium (Table 1). Reaction of S-phenylphenothiazinium hexafluorophosphate (2a) with phenyllithium (1a) in THF at −78 °C and 23 °C afforded biphenyl (3a) and N-methylphenothiazine (4) in good yields (entry 1). When the amount of 1a used was reduced, the conversion of 2a decreased (entry 2). In contrast, the reaction of S-phenylphenoxathiinium 5 with phenyllithium (1a) afforded a mixture of biphenyl (3a), phenoxathiin (6), and ring-opening product 7a (entry 3). As previously reported, the reaction of S-phenylthianthrenium 8 with phenyllithium (1a) under the same conditions afforded biphenyl (3a) in only 8% yield, while dibenzothiophene (9) (55%), diphenyl sulfide (10) (51%), and ring-opening product 7b (31%) were obtained in higher yields (entry 4).10 It was also reported that S-phenyldibenzothiophenium (11) afforded ring-opening product 7c in a quantitative yield (entry 5).11 It should be noted that ring opening product and N-methylcarbazole were not observed by 1H NMR analysis when 2a was used. These results suggest that the phenothiazine moiety is essential for successful formation of biaryl. The specificity of S-phenylphenothiazinium was examined by theoretical calculations, as described later.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Sulfonium | Products | |||
| Phenyllithium (1a) (0.6 mol) was added to sulfonium salt (0.2 mmol) in THF (5 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min and 23 °C for 1 h.a Determined by 1H NMR spectroscopy.b 0.4 mmol of 1a was used.c Isolated yield.d Result from ref. 11. | |||||
| 1 |

|

|

|
||
| 2a | 3a 91%a | 4 97%a | |||
| 2b | 2a | 3a 61%a | 4 60%a | ||
| 3 |

|

|

|

|
|
| 5 | 3a 38%a | 6 31%a | 7a 62%c | ||
| 4 |

|

|

|

|
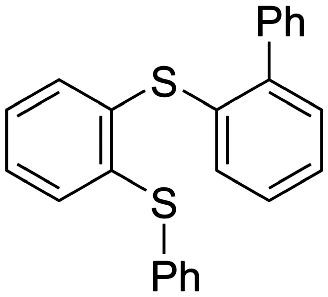
|
| 8 | 3a 8%a | 9 55%c | 10 51%c | 7b 31%c | |
| 5d |

|
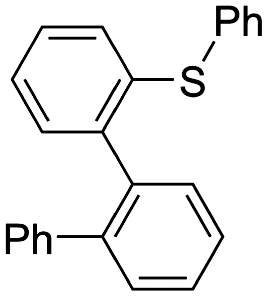
|
|||
| 11 | 7c quant. | ||||

The scope of the arylation of aryllithiums using S-arylphenothiazinium 2 investigated in the present study is shown in Scheme 1. To perform desymmetrizative mono-arylation of inexpensive 1,4-dibromobenzene, it was lithiated with n-BuLi and reacted with various S-arylphenothiaziniums successively to obtain the desired bromo-substituted biaryls (3b–3g) in good yields. Significantly, the reaction of S-(4-bromophenyl)phenothiazinium with aryllithiums bearing a bromo-substituent also afforded mono-arylated products bearing two bromo groups, 3h, 3i, and 3j. Other halogen-substituted biaryls could also be synthesized by the present method (3k–3o). The resulting bromo- or iodo-substituted biaryls are synthetically attractive intermediates for further derivatization by transition-metal-catalyzed cross-coupling reactions. It should be noted that bromo- or iodo-substituted biaryls have never been synthesized by Murahashi coupling because C–C bond formation takes place at the halogen-substituted position.4 Furthermore, various unsymmetrical biaryls were synthesized using the present method (3p–3z). Reactions using p-chlorophenyllithium produced the desired products in good yields (3p–3r), whereas the use of aryllithiums bearing a methylchalcogeno group slightly decreased the yields of the product (3s, 3t). Notably, the reaction efficiently produced 3p on the gram scale. Ortho- or meta-methyl- substituted aryllithium was applicable to the present reaction system (3u, 3v). Reactions of π-extended aryllithiums with S-arylphenothiaziniums also afforded the corresponding unsymmetrical biaryls in good yields (3w–3z).
 | ||
| Scheme 1 Scope of cross-coupling of aryllithiums and S-arylphenothiazinium ions for synthesizing biaryls. S-Arylphenothiazinium 2 (0.2 mmol) was added to a solution of aryllithium 1 (0.6 mmol) in THF (5 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 30 min and 23 °C for 1 h. Isolated yields of products are shown. See ESI,† for details. aThe reaction was performed on a gram scale. | ||
To gain insight into the selective formation of unsymmetrical biaryls from the S-arylphenothiazinium ions, density functional theory (DFT) calculations were conducted at the B3LYP/6-311G+(d,p) level. Possible reaction pathways of phenyllithium(THF)3 (1a) with S-phenylphenothiazinium (2a) via the corresponding sulfurane were explored. The relative free energy profiles of the formation of biphenyl (path A), ring-opening reaction (path B), and formation of diphenyl sulfide (path C) from tetracoordinated sulfuranes are summarized in Fig. 2a. Three types of sulfurane intermediates (IM A, IM B, and IM C) afforded the corresponding transition states (TS A, TS B, and TS C). Each sulfurane intermediate has a different pseudo-trigonal bipyramidal structure and similar free energy. Interconversion between IM A, IM B, and IM C occurs easily through pseudorotation.7a Therefore, the selectivity of the reaction products is determined by the TS, not by the relative stability of IM. TS A is energetically more stable than TS B and TS C, which is in good agreement with the experimental results, as described in Table 1.
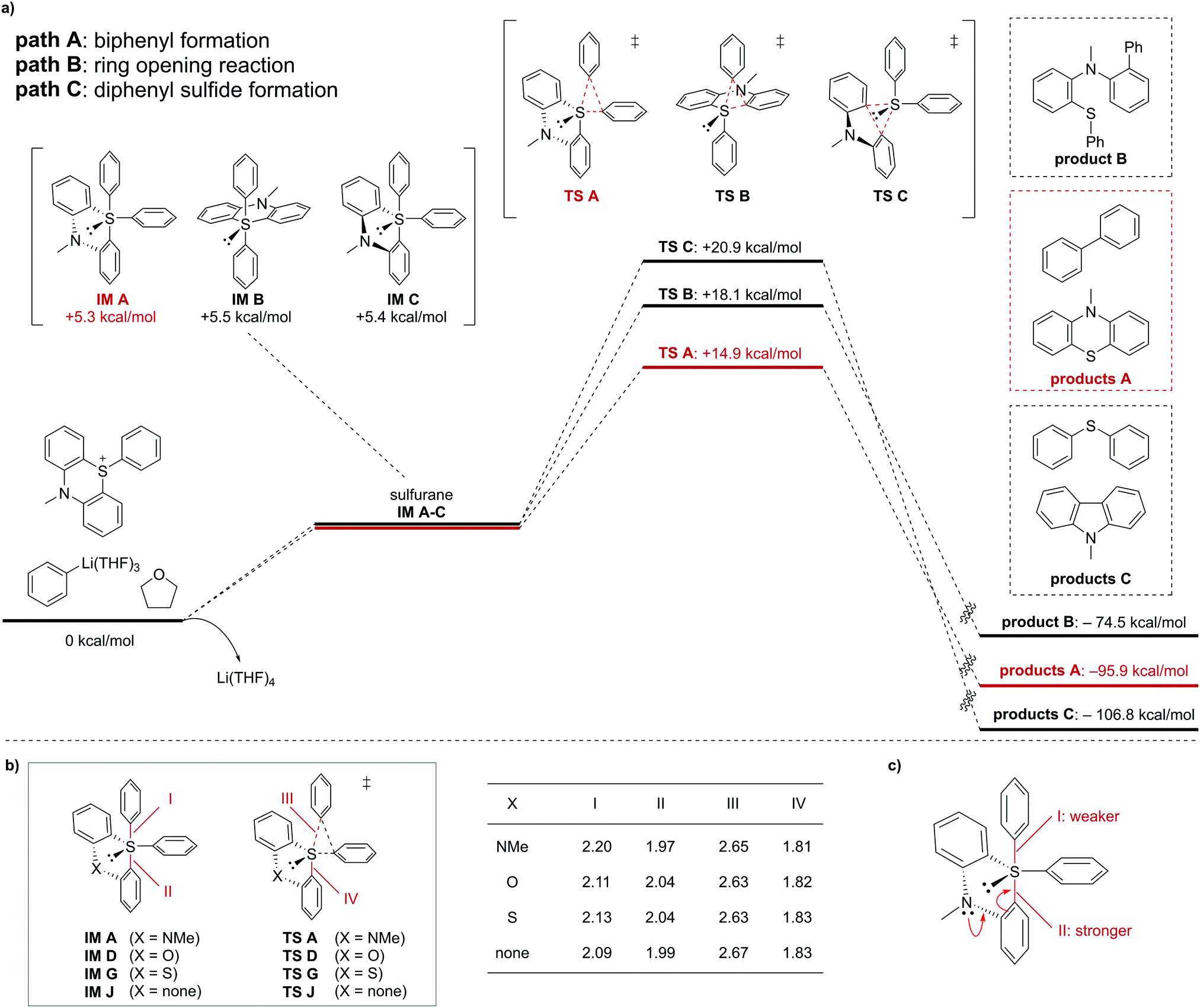 | ||
| Fig. 2 DFT calculations of reaction of phenyllithium (1a) and S-phenylphenothiazinium ion (2a) at the B3LYP/6-311G+(d,p) level. (a) Relative free energy profiles. (b) Selected calculated bond lengths (Å) or interatomic distances (Å) of sulfuranes and transition states for biphenyl formation. (c) A plausible reason for the structural difference in sulfurane IM A. | ||
DFT calculations using S-phenylphenoxathiinium (4), S-phenylthianthrenium (6), or S-phenyldibenzothiophenium (7) instead of 2a indicated that biphenyl formation was not energetically preferred to ring opening-reaction and formation of diphenyl sulfide when 4, 6, or 7 was used (see ESI,† for details). These computational results are consistent with the experimental results. To understand the major factor in the preference of path A for S-phenylphenothiazinium ion, we compared the structural differences between IM A and TS A with their analogs (Fig. 2b). The bond lengths of apical bonds III and IV in each transition state are almost the same. In contrast, apical bonds I and II in IM A are longer and shorter, respectively, than those in IM D, IM G, and IM J. IM A has a relatively close structure to TS A. This indicates that biphenyl formation from the S-arylphenothiazinium ion (path A) requires a smaller structural distortion with a lower energy loss than that from other sulfonium ions. Such a structural difference is due to π-electron donation by the lone pair on the nitrogen atom, which would strengthen and shorten apical C–S bond II (Fig. 2c). In general, when one apical bond is strengthened, the other apical bond becomes weaker and longer.12 Therefore, apical bond I in IM A is longer than that in others. This structural difference of IM A can be favorable for biaryl formation from phenyllithium (1a) and S-phenylphenothiazinium ion 2a.
In summary, we developed S-arylphenothiazinium ions as efficient arylation reagents for aryllithiums. The reaction can provide various unsymmetrical biaryls without transition-metal catalysis. Notably, transformable halogen substituents such as bromo- and iodo-groups are tolerated. The resulting halogen-substituted biaryls are attractive synthetic intermediates. This result contrasts sharply with that obtained through palladium-catalyzed Murahashi coupling. In contrast to other similar sulfonium salts, S-arylphenothiazinium ions are specifically suitable for biaryl synthesis through selective ligand coupling of a sulfurane intermediate, which is consistent with theoretical calculations. We are currently developing new transition-metal-free arylation reactions using S-arylphenothiazinium ions; the results will be presented in due course.
This research was partially supported by the MEXT-supported program for the Strategic Research Foundation at Private Universities, and the Society of Iodine Science. DFT calculations were performed using Research Center for Computational Science, Okazaki, Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. J. Wakefield, The Chemistry of Organolithium Compounds, Elsevier, Pergamon, 1974 Search PubMed.
- (a) N. Koike, T. Hattori, A. Takeda, Y. Okaishi and S. Miyano, Chem. Lett., 1997, 641 CrossRef CAS; (b) J.-M. Becht, A. Gissot, A. Wagner and C. Mioskowski, Chem. – Eur. J., 2003, 9, 3209 CrossRef CAS.
- (a) J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (b) F. Leroux and M. Schlosser, Angew. Chem., Int. Ed., 2002, 41, 4272 CrossRef CAS; (c) T. Truong, M. Mesgar, K. K. A. Le and O. Daugulis, J. Am. Chem. Soc., 2014, 136, 8568 CrossRef CAS.
- (a) S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408 CrossRef CAS; (b) A. Nagaki, A. Kenmoku, Y. Moriwaki, A. Hayashi and J.-I. Yoshida, Angew. Chem., Int. Ed., 2010, 49, 7543 CrossRef CAS; (c) M. Giannerini, M. Fananas-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS; (d) E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef; (e) D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354 CrossRef CAS.
- For selected examples of transition-metal-free biaryl-synthesis, see (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS; (b) T. Dohi, M. Ito, K. Morimoto, M. Iwata and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 1301 CrossRef CAS; (c) T. Morofuji, A. Shimizu and J.-I. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 7259 CrossRef CAS; (d) E. Shirakawa, Y. Hayashi, K.-I. Itoh, R. Watabe, N. Uchiyama, W. Konagaya, S. Masui and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 218 CrossRef CAS; (e) M. Ito, H. Kubo, I. Itani, K. Morimoto, T. Dohi and Y. Kita, J. Am. Chem. Soc., 2013, 135, 14078 CrossRef CAS; (f) T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582 CrossRef CAS; (g) M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Science, 2018, 362, 799 CrossRef CAS; (h) F. Kloss, T. Neuwirth, V. G. Haensch and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14476 CrossRef CAS; (i) A. Music, A. N. Baumann, P. Spieß, A. Plantefol, T. C. Jagau and D. Didier, J. Am. Chem. Soc., 2020, 142, 4341 CrossRef CAS.
- (a) W. A. Sheppard, J. Am. Chem. Soc., 1971, 93, 5597 CrossRef CAS; (b) S. Ogawa, Y. Matsunaga, S. Sato, I. Iida and N. Furukawa, J. Chem. Soc., Chem. Commun., 1992, 1141 RSC.
- (a) Ligand Coupling Reactions with Heteroatomic Compounds, ed. J.-P. Finet, Elsevier, Pergamon, 1998 Search PubMed; (b) Y. H. Khim and S. Oae, Bull. Chem. Soc. Jpn., 1969, 42, 1968 CrossRef CAS; (c) B. Trost, R. LaRochelle and R. Atkins, J. Am. Chem. Soc., 1969, 91, 2175 CrossRef; (d) R. W. LaRochelle and B. M. Trost, J. Am. Chem. Soc., 1971, 93, 6077 CrossRef CAS; (e) B. M. Trost and H. C. Arndt, J. Am. Chem. Soc., 1973, 95, 5288 CrossRef CAS; (f) S. Oae, Croat. Chem. Acta, 1986, 59, 129 CAS; (g) M. Hori, T. Kataoka, H. Shimizu, M. Ikemori and Y. Aoyama, J. Chem. Soc., Perkin Trans. 1, 1988, 1209 RSC; (h) S. Oae and Y. Uchida, Acc. Chem. Res., 1991, 24, 202 CrossRef CAS; (i) S. Oae, H. Ishihara and M. Yoshihara, Chem. Heterocycl. Compd., 1995, 31, 917 CrossRef; (j) S. Sato, O. Takahashi and N. Furukawa, Coord. Chem. Rev., 1998, 176, 483 CrossRef CAS; (k) W. M. Dean, M. Šiaučiulis, T. E. Storr, W. Lewis and R. A. Stockman, Angew. Chem., Int. Ed., 2016, 55, 10013 CrossRef CAS; (l) D. L. Chen, Y. Sun, M. Chen, X. Li, L. Zhang, X. Huang, Y. Bai, F. Luo and B. Peng, Org. Lett., 2019, 21, 3986 CrossRef CAS.
- (a) T. Kawai, N. Furukawa and S. Oae, Tetrahedron Lett., 1984, 25, 2549 CrossRef CAS; (b) N. Furukawa, T. Shibutani and H. Fujihara, Tetrahedron Lett., 1987, 28, 5845 CrossRef CAS; (c) M. Zhou, J. Tsien and T. Qin, Angew. Chem., Int. Ed., 2020, 59, 7372 CrossRef CAS.
- L. Racicot, T. Kasahara and M. A. Ciufolini, Org. Lett., 2014, 16, 6382 CrossRef CAS.
- B. Boduszek, H. J. Shine and T. K. Venkatachalam, J. Org. Chem., 1989, 54, 1616 CrossRef CAS.
- S. Sato and N. Furukawa, Tetrahedron Lett., 1995, 36, 2803 CrossRef CAS.
- L. J. Adzima, E. N. Duesler and J. C. Martin, J. Org. Chem., 1977, 42, 4001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, DFT calculations, and NMR data. See DOI: 10.1039/d0cc05830k |
| This journal is © The Royal Society of Chemistry 2020 |