
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titanium methyl tamed on silica: synthesis of a well-defined pre-catalyst for hydrogenolysis of n-alkane†
Aya
Saidi
 a,
Walid
Al Maksoud
a,
Manoja K.
Samantaray
*a,
Edy
Abou-Hamad
b and
Jean-Marie
Basset
*a
a,
Walid
Al Maksoud
a,
Manoja K.
Samantaray
*a,
Edy
Abou-Hamad
b and
Jean-Marie
Basset
*a
aKing Abdullah University of Science &Technology, Physical Science and Engineering, KAUST, Thuwal, 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa; manoja.samantaray@kaust.edu.sa
bImaging and Characterization Core Lab, King Abdullah University of Science and Technology, KAUST, Thuwal, 23955-6900, Saudi Arabia
First published on 5th October 2020
Abstract
Alkylation of Ti(CH3)2Cl21 by MeLi gives the homoleptic Ti(CH3)42 for the first time in the absence of any coordinating solvent. The reaction of 2 with silica pretreated at 700 °C (SiO2–700) gives two inequivalent silica-supported Ti-methyl species 3. Complex 3 was characterized by IR, microanalysis (ICP-OES, CHNS, and gas quantification), and advanced solid-state NMR spectroscopy (1H, 13C, DQ, TQ, and HETCOR). The catalytic activity of the pre-catalyst 3 is investigated in low-temperature hydrogenolysis of propane and n-butane with TONs of 419 and 578, respectively.
Conversion of inexpensive waxes, generated from the Fischer–Tropsch process to valuable chemicals/fuels, is a value-added process.1 Mainly, the hydrocracking process was used for the cracking of paraffinic waxes into useful chemicals.2,3 The primary obstacles in this process are the high temperature of reaction and the fast deactivation of the catalyst with the formation of coke.4 Many efforts were undertaken to reduce the temperature and carry out the conversion of waxes to valuable chemicals effectively.5 Since the discovery of the well-defined silica-supported Zr hydride, [(
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O)3ZrH], which was able to catalyze the hydrogenolysis of a given alkane to lower homologs and the de-polymerization (Ziegler–Natta de-polymerization) of LLDPE to fuel range alkanes at relatively low temperature (150 °C), a new door was opened up for research in this new field.6,7
Si–O)3ZrH], which was able to catalyze the hydrogenolysis of a given alkane to lower homologs and the de-polymerization (Ziegler–Natta de-polymerization) of LLDPE to fuel range alkanes at relatively low temperature (150 °C), a new door was opened up for research in this new field.6,7
Recently, we observed that moving from metal neopentyl to metal methyl, the reactivity in alkane metathesis reaction increased several-fold.8 Additionally, it was comparatively easy to characterize the catalyst and understood the possible reaction mechanism by either DFT or by isolating the active intermediate by isotopically labeling the methyls attached to metal center.9,10 In this regard, we utilized various homoleptic metal methyl homogeneous complexes, which are very rare, very reactive in the homogeneous phase, and almost not suitable for catalysis because of their unstable nature at room temperature.11–14 To make them stable for catalysis, we used Surface Organometallic Chemistry (SOMC) approach, which stabilizes molecular species otherwise unstable in solution. Metal alkyl can react with surface silanols and form a stable catalyst at room temperature, which makes them comparatively easier to use for catalysis.11,15
Herein, we disclose for the first time the synthesis and characterization of a homoleptic Ti(CH3)4 complex, its grafting on silica partially dehydroxylated at 700 °C (SiO2–700), characterization of the grafted material, and its application in hydrogenolysis of propane and n-butane.
Synthesis of an ether bound (Et2O)Ti(CH3)4 without sufficient proof is already known in the literature,16,17 but for our catalytic purposes, we wanted a homoleptic Ti without any coordination of the solvent as the coordinated solvent makes them less electropositive and eventually less effective for C–H bond activation. The difficulty in preparing such metal complexes is already reported in the literature,18,19 only partial methylated species of metals of group 4 are known so far.20–22 Therefore, we choose a soluble metal precursor (TiCl4) and a non-coordinated solvent (pentane and dichloromethane) for this reaction. It appeared that the reaction was not straightforward and required two steps (Scheme 1).
 | ||
| Scheme 1 Synthesis of TiCl2(CH3)21 and Ti(CH3)42. | ||
In the first step, the TiCl4 precursor is reacted with one equivalent of Zn(CH3)2 in a mixture of pentane and dichloromethane (1/1) at a temperature range between −80 °C to −40 °C for 4 hours to yield an orange solution of TiCl2(CH3)2.16 In the end, the reaction mixture was filtered, and the filtrate was dried under vacuum. An aliquot of TiCl2(CH3)2 was taken in an NMR tube and was mixed with CD2Cl2 and analyzed by liquid-state NMR (1H, 13C, HSQC, and 35Cl) at −40 °C. 1H and 13C NMR spectra (Fig. S1A and B, ESI†) display peaks at 2.6 ppm and 99.1 ppm, respectively, attributed to the hydrogen and carbon atoms of the methyl group of Ti–CH3. This was further proved by the correlation between both of the peaks observed in the HSQC spectrum (Fig. S1C, ESI†). Additionally, 35Cl NMR was conducted, and a peak at 601.1 ppm confirms the presence of Ti–Cl (Fig. S1D, ESI†).23–25
In the second step, the TiCl2(CH3)2 reacted with an excess of MeLi (50% enriched) in pentane at a temperature range between −80 °C to −60 °C for 3 hours, affording a red color solution of the desired product Ti(CH3)4 (Scheme 1). The complex is highly sensitive to oxygen, moisture, and temperature; black fumes were observed immediately when the temperature was increased above −60 °C, which makes it very difficult to isolate the complex. Therefore, at the end of the reaction, the reaction mixture is filtered and used immediately for grafting on oxide surfaces.
In an NMR tube, 0.2 mL of TiCl4 mixed with one equivalent of ZnMe2, pentane, and CD2Cl2 at −80 °C and let the reaction to run for 1 hour, then an excess of 13C enriched MeLi was added to it, and the reaction continued in the NMR tube for another 3 hours at −60 °C. The progress of the reaction was analyzed by liquid-state NMR (1H, 13C, and 35Cl). In the 1H and 13C NMR spectra (Fig. 1A and B), we observed that the peaks of the 1H and 13C were shifted from 2.6 and 99.1 ppm (as observed in the case of TiCl2(CH3)2) to 1.99 and 72.9 ppm, respectively, for the newly formed Ti(CH3)4 complex. This is understandable as complete alkylation occurs on the Ti center causes an upfield shift of the carbon and proton peaks of methyl ligands. The attribution of the obtained chemical shifts in the liquid-state NMR is realized by comparing them with those of the reported ether bound methyl titanium chlorides.16,26
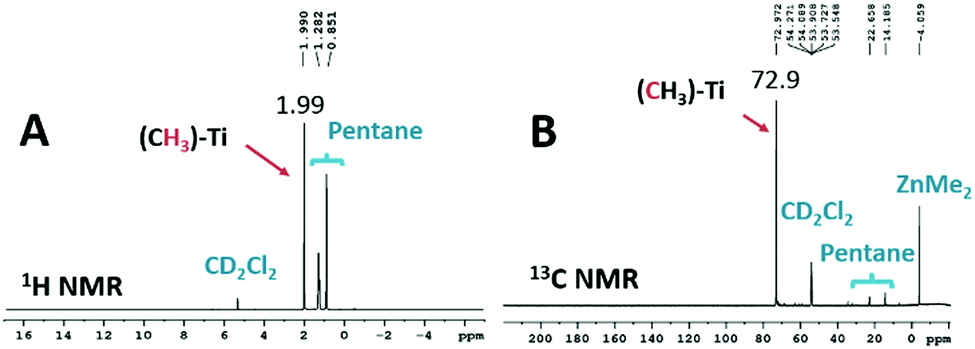 | ||
| Fig. 1 (A) 1H and (B) 13C NMR spectra of Ti(CH3)4 in CD2Cl2 at 213 K. | ||
To confirm the complete substitution of chlorine atom by methyl ligand, we characterize the sample by 35Cl NMR (Fig. S2, ESI†). The 35Cl spectrum of the complex does not show any chlorine peak (which usually comes in between 500–800 ppm depending upon the number of chlorine atoms attached to Ti23,24,27) except that of the deuterated solvent CD2Cl2. This further corroborates the successful synthesis of a homoleptic Ti(CH3)4 complex without any coordinated solvent.
Grafting of Ti(CH3)4 was performed in situ by reacting an excess of 2 with partially dehydroxylated silica (silica pretreated at 700 °C, SiO2–700) at −60 °C in pentane under an inert atmosphere of argon for 4 hours (Scheme 2), followed by washing with pentane three times to remove the excess of 2 before drying under high vacuum (10−5 mbar). In the end, a dark brown powder was obtained. The grafting process generates the formation of two environmentally different [(Si–O–)TiMe3] species resulting in the interaction with the silanol groups and Si–O–Si bridges (Scheme 2). As expected, from previous work carried out with WMe6/silica, once grafted, the Ti complex is much more stable and prone to more in-depth characterization.28,29
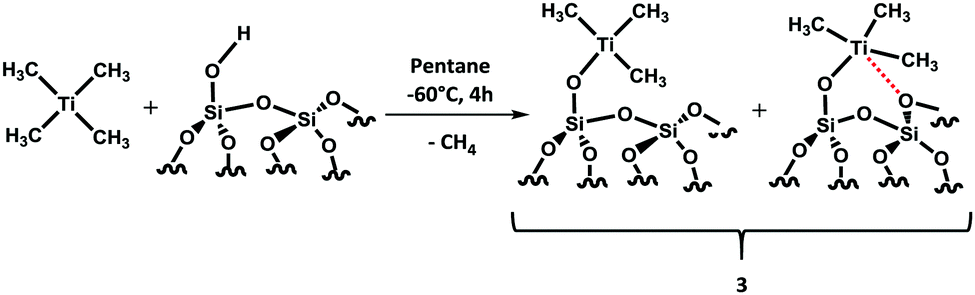 | ||
| Scheme 2 Reaction of Ti(CH3)4 with silica-700 at −60 °C. | ||
The grafted complex 3 was characterized by FT-IR spectroscopy, elemental analysis, methane quantification, and solid-state NMR spectroscopy (1H, 13C, DQ, TQ, and HETCOR). The FTIR spectrum was recorded at 25 °C (Fig. S3, ESI†); the IR peak of the isolated silanols at 3747 cm−1 completely disappeared, and groups of new bands appeared in 3005–2819 and 1454 cm−1 region. These bands are respectively assigned to the ν(CH) and δ(CH) vibrations of the methyl ligands coordinated to titanium. Elemental analysis of 3 was conducted and it gave 1.8% Ti, 1.3% C and 0.33% H, with a ratio of C/Ti = 2.88 and H/Ti = 8.77. These results further prove the formation of complex 3.
Furthermore, the structure of 3 was characterized by solid-state NMR spectroscopy. The 1H magic-angle spinning (MAS) NMR spectrum of 3 displays one signal at 2.0 ppm (Fig. 2A) that autocorrelated on the diagonals of both double-quantum (DQ) and triple-quantum (TQ) NMR spectra at 4.0 and 6.0 ppm, respectively (Fig. 2B and C) which proves that they refer to –CH3 group. The 13C cross-polarization magic-angle spinning (CP-MAS) NMR spectrum shows a main peak at 76 ppm along with a small signal at 61 ppm (Fig. 2D) that correlate with the proton at 2 ppm as indicated in the 2D 1H–13C HETCOR NMR spectrum (Fig. 2E). These peaks are assigned to the carbon of the methyl of the titanium species 3 (both with and without interaction with the oxygen atom of the siloxane bridge). This type of interaction was already reported by our group with well-defined [(Si–O–)TaCl2Me2] species.29 The 13C signal in solid-state NMR is slightly shifted compared to the carbon peak in the solution NMR spectrum from 72.9 to 76 ppm; this is understandable as we have replaced at least one methyl group with an electronegative oxygen atom.
 | ||
| Fig. 2 Solid-state NMR characterization of grafted Ti(CH3)4 onto SiO2-700; (A) one-dimensional (1D) 1H MAS solid-state NMR spectrum of 3 (acquired on a 600 MHz NMR spectrometer (14.1 T) with a 22 kHz MAS spinning frequency, a repetition delay of 5 s, and 8 scans), (B) two-dimensional (2D) 1H–1H double-quantum (DQ) spectrum of 3, (C) two-dimensional (2D) 1H–1H triple-quantum (TQ) spectrum of 3 (acquired on a 600 MHz NMR spectrometer at 22 kHz MAS spinning frequency with a back-to-back recoupling sequence, number of scans 128, repetition delay 5 s, number of t1 increments 128), (D) 13C CP/MAS NMR spectrum of 3 (acquired on a 400 MHz NMR spectrometer (9.4 T) with a 10 kHz MAS frequency, 20 K scans, a 4 s repetition delay, and a 2 ms contact time. Exponential line broadening of 80 Hz was applied before Fourier transformation), (D) 2D 1H–13C CP/MAS dipolar HETCOR spectrum of 3 (acquired on a 400 MHz NMR spectrometer (9.4 T) with a 10 kHz MAS frequency, 2048 scans per t1 increment, a 4 s repetition delay, 64 individual t1 increments and a 0.2 ms contact time). | ||
Additionally, the structure of 3 was confirmed by the gas quantification method after hydrolysis. The hydrolysis reaction of 50 mg of complex 3 using degassed water produced 0.937 mmol g−1 of methane; theoretically, we should get 1.065 mmol g−1, which is consistent with the formation of [(SiO–)TiMe3] surface species.
After the characterization of complex 3, it was tested for hydrogenolysis of propane and n-butane. In a typical experiment, complex 3 (0.088 mmol of Ti) was loaded in a continuous flow reactor, and a mixture of propane or n-butane (3.5 mL min−1) and hydrogen (9 mL min−1) was passed through the catalytic bed at 180 °C. All the reactions were conducted at the atmospheric pressure (P = 1 bar). The conversion of the reactant and the gas products were analyzed every 20 min by on-line gas chromatography.
We achieved a maximum conversion of 85.9% after 40 min and a TON of 419 for propane hydrogenolysis and maximum conversion of 99.8% and a TON of 578 for n-butane hydrogenolysis at our contact time (Fig. 3A and B).
 | ||
| Fig. 3 Evolution of conversion (blue curve) and corresponding TON over time (red curve) for hydrogenolysis reaction of (A) propane (B) n-butane using 3. Evolution of product selectivities over time for hydrogenolysis reaction of (C) propane (D) n-butane using 3. | ||
During the hydrogenolysis reaction of propane, we observed that the selectivities of methane and ethane remain similar (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) (Fig. 3C), which corroborates the previously reported results that β-methyl transfer is only possible in the case of Ti.30 whereas, in the case of n-butane, methane, ethane, and propane (Fig. 3D) were observed. From the previously reported data, we know that hydrogenolysis can take place either by α-alkyl or β-alkyl migration, depending on the metal atom.31,32 Our results indicate that in our catalytic system, we only observe β-alkyl transfer (as we observed ethane, propane, along with methane instead of only methane) followed by the formation of Ti(carbene)(alkyl) intermediate and finally, hydrogenolysis to form lower alkanes (Scheme S1, ESI†). In the beginning, we observed that the selectivity for ethane is higher than other products, and ethane was considered to be the primary product. However, with time on stream, the evolution of methane and propane occurs. At the end of the reaction, we observed that the selectivity for propane, methane, and ethane remains similar. It is surprising to see that the selectivity for propane is similar to that observed for ethane and methane. We believe this is because of the low contact time of the reactant to the catalyst, and the concentration of the propane is much less to react back with the catalyst.
:50) (Fig. 3C), which corroborates the previously reported results that β-methyl transfer is only possible in the case of Ti.30 whereas, in the case of n-butane, methane, ethane, and propane (Fig. 3D) were observed. From the previously reported data, we know that hydrogenolysis can take place either by α-alkyl or β-alkyl migration, depending on the metal atom.31,32 Our results indicate that in our catalytic system, we only observe β-alkyl transfer (as we observed ethane, propane, along with methane instead of only methane) followed by the formation of Ti(carbene)(alkyl) intermediate and finally, hydrogenolysis to form lower alkanes (Scheme S1, ESI†). In the beginning, we observed that the selectivity for ethane is higher than other products, and ethane was considered to be the primary product. However, with time on stream, the evolution of methane and propane occurs. At the end of the reaction, we observed that the selectivity for propane, methane, and ethane remains similar. It is surprising to see that the selectivity for propane is similar to that observed for ethane and methane. We believe this is because of the low contact time of the reactant to the catalyst, and the concentration of the propane is much less to react back with the catalyst.
To our knowledge, we synthesized the first homoleptic Ti(CH3)4 complex without any coordinating solvent. The homogeneous complex Ti(CH3)4 is very reactive and starts to decompose at a temperature above −60 °C, which makes this complex very difficult to study and use for any catalytic reaction. For the first time, it was stabilized on partially dehydroxylated silica (SiO2–700) and was characterized at the molecular level using advanced solid-state NMR, IR and, gas quantification methods. The well-defined silica-supported pre-catalyst 3 was used for hydrogenolysis of propane and n-butane with a TON of 419 and 578, respectively. Other aspects of this complex are under study in our laboratory.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Bouchy, G. Hastoy, E. Guillon and J. A. Martens, Oil Gas Sci. Technol., 2009, 64, 91–112 CrossRef CAS.
- C. Gambaro, V. Calemma, D. Molinari and J. Denayer, AIChE J., 2011, 57, 711–723 CrossRef CAS.
- J. Lee, S. Hwang, J. G. Seo, S. B. Lee, J. C. Jung and I. K. Song, J. Ind. Eng. Chem., 2010, 16, 790–794 CrossRef CAS.
- J. W. Gosselink and W. H. J. Stork, Ind. Eng. Chem. Res., 1997, 36, 3354–3359 CrossRef CAS.
- T. Hanaoka, T. Miyazawa, K. Shimura and S. Hirata, Catalysts, 2015, 5, 1983–2000 CrossRef CAS.
- J. Corker, F. Lefebvre, C. Lecuyer, V. Dufaud, F. Quignard, A. Choplin, J. Evans and J. M. Basset, Science, 1996, 271, 966–969 CrossRef CAS.
- V. R. Dufaud and J. M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef CAS.
- M. K. Samantaray, E. Callens, E. Abou-Hamad, A. J. Rossini, C. M. Widdifield, R. Dey, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2014, 136, 1054–1061 CrossRef CAS.
- Y. Chen, E. Abou-hamad, A. Hamieh, B. Hamzaoui, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2015, 137, 588–591 CrossRef CAS.
- N. Maity, S. Barman, E. Callens, M. K. Samantaray, E. Abou-Hamad, Y. Minenkov, V. D'Elia, A. S. Hoffman, C. M. Widdifield, L. Cavallo, B. C. Gates and J. M. Basset, Chem. Sci., 2016, 7, 1558–1568 RSC.
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J. M. Basset, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC.
- M. K. Samantaray, V. D'Eia, E. Pump, L. Falivene, M. Harb, S. O. Chikh, L. Cavallo and J. M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS.
- M. K. Samantaray, S. Kavitake, N. Morlanes, E. Abou-Hamad, A. Hamieh, R. Dey and J. M. Basset, J. Am. Chem. Soc., 2017, 139, 3522–3527 CrossRef CAS.
- M. K. Samantaray, R. Dey, S. Kavitake, E. Abou-Hamad, A. Bendjeriou-Sedjerari, A. Hamieh and J. M. Basset, J. Am. Chem. Soc., 2016, 138, 8595–8602 CrossRef CAS.
- J. M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS.
- S. Kleinhenz and K. Seppelt, Chem. – Eur. J., 1999, 5, 3573–3580 CrossRef CAS.
- H. Berthold and G. Groh, Z. Anorg. Allg. Chem., 1963, 319, 230–235 CrossRef CAS.
- H. Makio, T. Oshiki, K. Takai and T. Fujita, Chem. Lett., 2005, 34, 1382–1383 CrossRef CAS.
- H. Makio, T. Ochiai, J.-i. Mohri, K. Takeda, T. Shimazaki, Y. Usui, S. Matsuura and T. Fujita, J. Am. Chem. Soc., 2013, 135, 8177–8180 CrossRef CAS.
- Y. Gao, M. D. Christianson, Y. Wang, J. Chen, S. Marshall, J. Klosin, T. L. Lohr and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 7822–7830 CrossRef CAS.
- I. E. Nifant’ev, P. V. Ivchenko, V. V. Bagrov, S. M. Nagy, S. Mihan, L. N. Winslow and A. V. Churakov, Organometallics, 2013, 32, 2685–2692 CrossRef.
- D. Balboni, I. Camurati, A. C. Ingurgio, S. Guidotti, F. Focante and L. Resconi, J. Organomet. Chem., 2003, 683, 2–10 CrossRef CAS.
- M. A. Fedotov, O. L. Malkina and V. G. Malkin, Chem. Phys. Lett., 1996, 258, 330–335 CrossRef CAS.
- A. J. Rossini, R. W. Mills, G. A. Briscoe, E. L. Norton, S. J. Geier, I. Hung, S. Zheng, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2009, 131, 3317–3330 CrossRef CAS.
- Y. Saito, Can. J. Chem., 1965, 43, 2530–2534 CrossRef CAS.
- S. Berger, W. Bock, G. Frenking, V. Jonas and F. Mueller, J. Am. Chem. Soc., 1995, 117, 3820–3829 CrossRef CAS.
- E. S. Blaakmeer, F. J. Wensink, E. R. H. van Eck, G. A. de Wijs and A. P. M. Kentgens, J. Phys. Chem. C, 2019, 123, 14490–14500 CrossRef CAS.
- M. K. Samantaray, R. Dey, E. Abou-Hamad, A. Hamieh and J. M. Basset, Chem. – Eur. J., 2015, 21, 6100–6106 CrossRef CAS.
- Y. Chen, E. Callens, E. Abou-Hamad, N. Merle, A. J. P. White, M. Taoufik, C. Coperet, E. Le Roux and J. M. Basset, Angew. Chem., Int. Ed., 2012, 51, 11886–11889 CrossRef CAS.
- C. Rosier, G. P. Niccolai and J. M. Basset, J. Am. Chem. Soc., 1997, 119, 12408–12409 CrossRef CAS.
- S. Norsic, C. Larabi, M. Delgado, A. Garron, A. de Mallmann, C. Santini, K. C. Szeto, J. M. Basset and M. Taoufik, Catal. Sci. Technol., 2012, 2, 215–219 RSC.
- V. Polshettiwar, F. A. Pasha, A. De Mallmann, S. Norsic, J. Thivolle-Cazat and J. M. Basset, ChemCatChem, 2012, 4, 363–369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05816e |
| This journal is © The Royal Society of Chemistry 2020 |