
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical control of photoinduced charge-transfer direction in a tetrathiafulvalene-fused dipyrrolylquinoxaline difluoroborate dyad†
Ping
Zhou
a,
Ulrich
Aschauer
 *a,
Silvio
Decurtins
a,
Thomas
Feurer
b,
Robert
Häner
a and
Shi-Xia
Liu
*a
*a,
Silvio
Decurtins
a,
Thomas
Feurer
b,
Robert
Häner
a and
Shi-Xia
Liu
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: ulrich.aschauer@dcb.unibe.ch; liu@dcb.unibe.ch
bInstitute of Applied Physics, University of Bern, Sidlerstrasse 5, CH-3012 Bern, Switzerland
First published on 5th October 2020
Abstract
A new approach for a compact annulation of tetrathiafulvalene (TTF) and dipyrrolylquinoxaline difluoroborate (QB) is presented, leading to strong electronic interactions between the TTF and QB units. Regulation of distinct photoinduced charge flows within this dyad is achieved by external stimuli, which is also verified by TD-DFT calculations.
Inspired by the unidirectionality of electron transfer evidenced in photosystem II, special attention has been devoted to efficient control of electron flow in π-conjugated ensembles consisting of electron donor (D) and acceptor (A) subunits.1 However, regulating the direction of intramolecular charge transfer (ICT) over multiple pathways present in such D–A systems remains a big challenge.2 An in-depth understanding of this key issue is a prerequisite for the successful implementation of organic molecules in photovoltaics, electronics and photonics.3 Their intrinsic electronic properties are governed by the extent of communication between D and A units which can be fine-tuned by the meticulous choice and modification of the D–A architectures as well as external stimuli such as coordination,4 protonation,5 redox6,7 and light.8 Very recently, a fullerene–phenothiazine dyad was reported to show in situ switchable molecular photodiode-like behaviour by regulating the redox state of phenothiazine.7 Our strategy involves a direct annulation of a redox-active π-electron donor TTF9 (black part, Chart 1) and a strong chromophore dipyrrolylquinoxaline difluoroborate (QB, blue part, Chart 1) leading to the formation of the TTF–QB dyad. QB is analogous to BODIPY (boron dipyrromethene) with high extinction coefficient and fluorescence quantum yield and can act as a π-electron acceptor.10 In stark contrast to the previously reported TTF–BODIPY ensembles,11 TTF–QB absorbs strongly in the visible spectral region due to a strong effective ICT dominated by one-electron excitation from the HOMO localized on the TTF moiety to the LUMO on the QB core, indicative of largely enhanced electronic communication between them. On the other hand, TTF–QB featured with a peripheral pyrrolic NH unit, is envisaged to form a hydrogen bond with a fluoride, leading to an ICT from the HOMO mainly localized on the pyrrolylquinoxaline–fluoride adduct to the LUMO on the pyrrolylquinoxaline difluoroborate coordination moiety. Moreover, upon chemical oxidation of the TTF subunit to its radical cation TTF˙+, a reverse ICT from the QB to the TTF moiety would occur. As a consequence, chemical regulation of photoinduced charge flow within this compact multicomponent dyad is fulfilled.
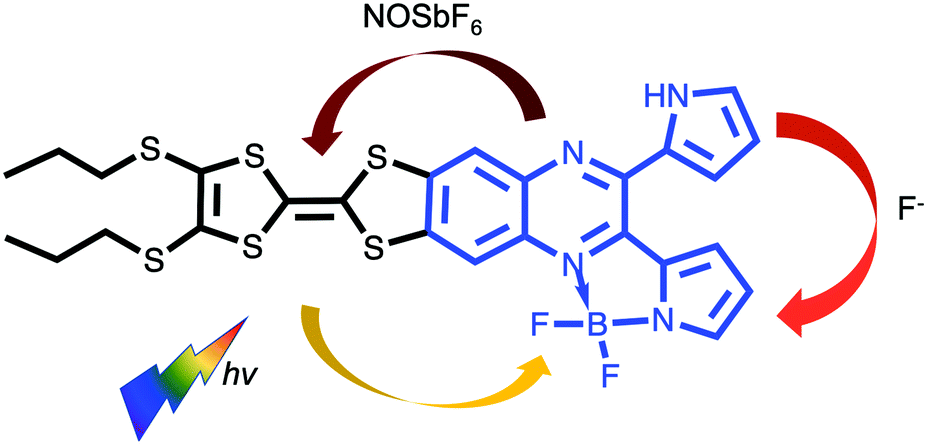 | ||
| Chart 1 Chemical structure of the target dyad TTF–QB and chemical regulation of ICT direction in the presence of fluoride and oxidant NOSbF6. | ||
In the present contribution, the synthesis and electronic properties of TTF–QB in response to external stimuli including H-bonded fluoride adduct and chemical oxidation are discussed.
The target dyad TTF–QB was readily prepared in a yield of 60% via a reaction of a TTF-fused 2,3-di(1H-2-pyrrolyl)quinoxaline (TTF–PQ)12 with BF3OEt2 using DBU as a base (Scheme S1, ESI†). A coplanar conformation of the resultant pyrrolide with the quinoxaline core renders a chelating ligand forming a stable five-membered ring with a boron atom, which leads to extended π-conjugation in the dyad TTF–QB. Purification by chromatographic separation on silica gel afforded the analytically pure product which was fully characterized. 1H NMR and mass spectrometric data match well with its predicted molecular structure.
The electrochemical properties of TTF–QB, the free ligand TTF–PQ and the reference compounds dipyrrolylquinoxaline (PQ) or QB in CH2Cl2 were investigated by cyclic voltammetry (Fig. S1, S2 and Table S1, ESI†). TTF–QB undergoes two reversible oxidation processes at 0.78 and 1.10 V to the TTF radical cation and dication states and one reversible reduction process at −0.81 V, corresponding to the reduction of the QB subunit. Upon complexation of the PQ unit with one equivalent of BF2, both oxidation and reduction potentials are positively shifted, indicative of the enhanced π-electron withdrawing ability of the quinoxaline core due to B–N bond formation between the nitrogen lone pair and the vacant p-orbital of the boron atom. It can therefore be deduced that the resultant QB subunit acts as a stronger π-electron acceptor than the quinoxaline unit itself. A separation between the onset oxidation and reduction potentials leads to an electrochemical HOMO–LUMO gap of 1.37 eV for TTF–QB, which is significantly reduced compared to that for TTF–PQ (1.91 eV).
The dark blue TTF–QB strongly absorbs over an extended range within the UV-Vis spectral region (Fig. 1). A strong and broad absorption band (ε ≈ 2.3 × 104 M−1 cm−1) peaked at 610 nm is attributed to an ICT transition from the TTF unit to the QB fragment as confirmed theoretically (vide infra). A series of less-intense absorption bands is observed between 550 nm and 350 nm, followed by a very intense absorption band centered at 310 nm. These spectral features are in stark contrast with those of the free ligand TTF–PQ. On the one hand, its lowest energy absorption band is hypsochromically shifted by 120 nm, as evidenced by its purple-red colour. This observation is attributed to a weaker electron-withdrawing effect of the quinoxaline itself than the QB subunit. Consequently, the optical HOMO–LUMO gap of TTF–PQ (2.23 eV) is much larger than that of TTF–QB (1.69 eV). On the other hand, its absorption spectrum looks much simpler due to the decreased π-conjugation of the molecule induced by the non-planarity of two pyrrole rings with the quinoxaline core. In other words, BF2 complexation leads to an extended π-conjugation, which renders a series of electronic transitions observable in the visible spectral region. This assumption is corroborated by the computational results (vide infra). All these spectroscopic results are in good agreement with the aforementioned electrochemical properties.
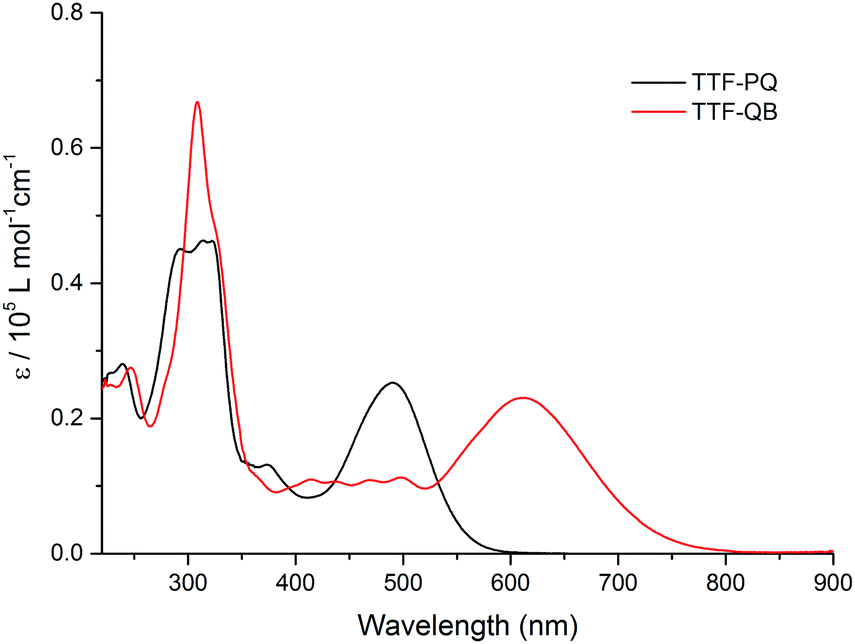 | ||
| Fig. 1 The UV-Vis absorption spectra of TTF–PQ and TTF–QB (1.6 × 10−5 M) in CH2Cl2 at room temperature. | ||
A successive addition of tetrabutylammonium fluoride trihydrate (TBAF) in CH2Cl2 leads to a profound change in the absorption spectrum of TTF–QB (Fig. 2). The occurrence of clearly defined isosbestic points at 345 nm, 530 nm and 655 nm indicates the conversion of TTF–QB to its corresponding H-bonded species with F−. The low-energy ICT absorption band decreases at the benefit of a new ICT transition at 650 nm which is bathochromically shifted by 40 nm in good agreement with a significant colour change from blue to yellow-green. This behaviour is ascribed to the formation of N–H⋯F hydrogen bonds, as previously observed during the titration of the ligand TTF–PQ with the fluoride anion.12 Concomitantly, the emergence of a strong absorption band at 410 nm occurs at the expense of the absorption band around 310 nm. Upon addition of 80 equiv. of TBAF, no further noticeable spectral changes are observed. The final spectrum resembles that of the reference QB in the presence of 80 equiv. TBAF, by exhibiting three distinct electronic transitions (Fig. S4, ESI†). As expected, all these transitions are bathochromically shifted and much stronger in TTF–QB than in QB, by virtue of the large extended π-conjugation that substantially lowers the energy level of the LUMO. In the following, quantum-chemical calculations demonstrate that the lowest energy transition of TTF–QB, when H-bonded to fluoride, corresponds to an ICT from the pyrrole⋯F− unit to the QB moiety, suggesting that the direction of the ICT in TTF–QB can be regulated by adding F−.
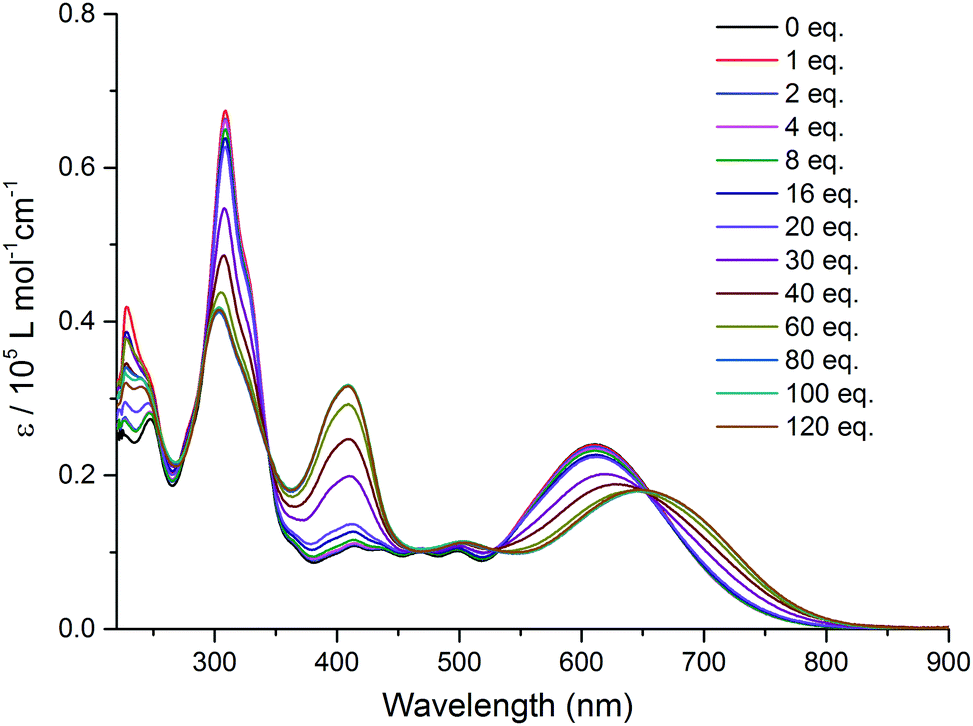 | ||
| Fig. 2 Variation of UV-Vis absorption spectra of TTF–QB (1.5 × 10−5 M) in CH2Cl2 upon successive addition of aliquots of TBAF at room temperature. | ||
Intramolecular electronic interactions between the TTF and QB units in the TTF–QB dyad have further been explored by UV-Vis-NIR spectroscopy, whereby a chemical oxidation of TTF–QB was carried out by successive addition of NOSbF6 aliquots at room temperature. As depicted in Fig. 3, a progressive reduction of the absorbance of both π–π* and ICT transitions at 310 nm and 610 nm, respectively, is accompanied with a concomitant emergence of new absorption bands at 736 nm and around 1040 nm which reach their maximum values upon addition of 2.5 equiv. of NOSbF6. These new transitions are characteristic of the TTF radical cation species TTF˙+ within a D–A ensemble.13 We note that in previously reported studies on oxidised D–A compounds,14 the lowest energy absorption band is assigned to the dimeric radical cation species arising from enforced intermolecular interactions. In the actual multicomponent molecule, however, the two lowest energy electronic transitions in the NIR around the asymmetric 1040 nm absorbance show a back CT character, which is corroborated by TD-DFT calculations in the following.
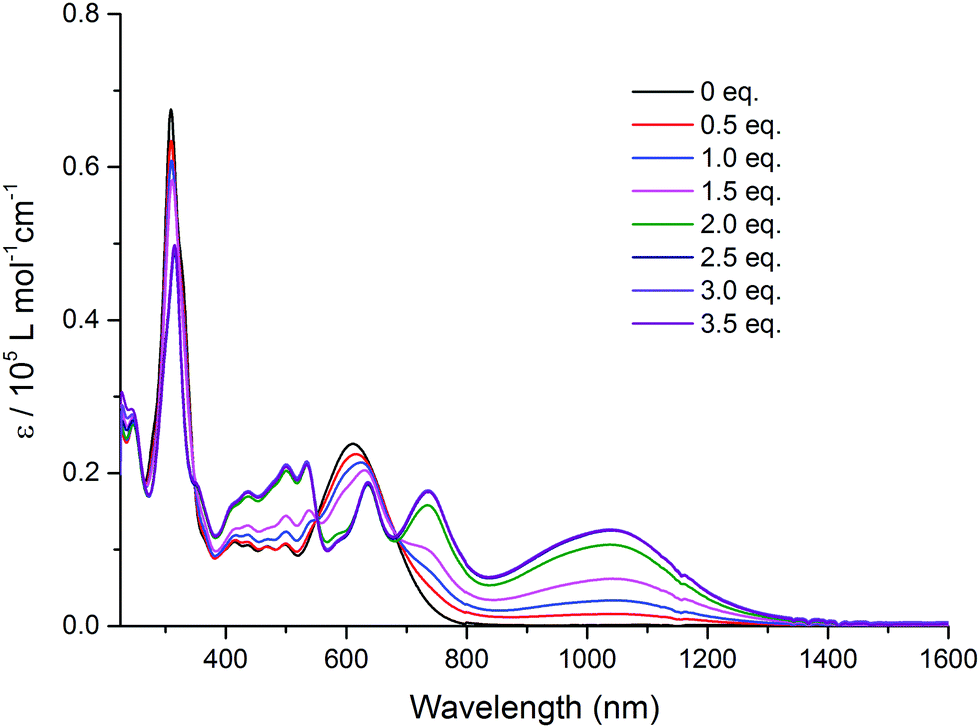 | ||
| Fig. 3 Variation of UV-Vis-NIR absorption spectra of TTF–QB (1.5 × 10−5 M) in CH2Cl2 upon successive addition of aliquots of NOSbF6 at room temperature. | ||
To characterise and verify the various electronic transitions, TD-DFT calculations were accomplished using the Gaussian 1615 package at the B3LYP/6-31G(d,p) level of theory. After DFT relaxation to within default thresholds, TD-DFT calculations of the 40 lowest excited states were carried out and the absorption spectra extracted with GaussSum 3.0.16 The predicted absorption spectra of TTF–QB, its fluoride adduct and its oxidised form are in fairly good agreement with experimental results (Tables S2–S7 and Fig. S11, ESI†). Compared to TTF–PQ, TTF–QB reveals in particular an absorption onset at a much larger wavelength. This lowest energy absorption band, peaked at 610 nm, is attributed to the S0 → S1 excitation, almost exclusively (99%) dominated by the HOMO–LUMO transition that involves CT from the TTF to the QB moiety, as clearly shown by the associated molecular orbitals in Fig. 4a. This directionality of the longest wavelength ICT is a kind of standard for fused TTF–acceptor ensembles.13 Similarly, also the calculated excitations at higher energy match well with the observed absorption spectrum. In the actual case of the fluoride adduct, however, the fluoride binding on the acceptor side destabilises the occupied MO localised on the QB site such that it represents now the HOMO of the dyad (Fig. 4b). Consequently, the corresponding HOMO–LUMO transition (96% of the calculated S0 → S1 excitation) corresponds to a charge flow predominantly in an orthogonal direction and at longer wavelength compared to the former one, in full agreement with the observed bathochromic shift of the ICT band by 40 nm. The latter energy shift is well substantiated by the calculation. Also in the blue/UV spectral region, the calculated excitations represent closely the experimental absorption bands. Upon oxidation to the radical cation species TTF˙+–QB, a broad and clearly asymmetric absorption feature emerges in the NIR region at wavelengths above 800 nm, which points to two new excitations. In accordance with it, the calculation of the open-shell molecule reveals two electronic transitions in the NIR part, D0 → D1 (98% β-HOMO to β-LUMO) and D0 → D2 (97% β-HOMO−1 to β-LUMO) at 1483 nm and 1075 nm, respectively. Importantly, both excitations reveal that upon photoexcitation, the charge flow is directed in the reverse direction, hence from the QB to the TTF˙+ unit which is now the acceptor site (Fig. 4c, Tables S6 and S7, ESI†). Moreover, the calculated excitations D0 → Dn (n ≥ 3) cover well the experimental absorption features at wavelengths around 800 nm and lower.
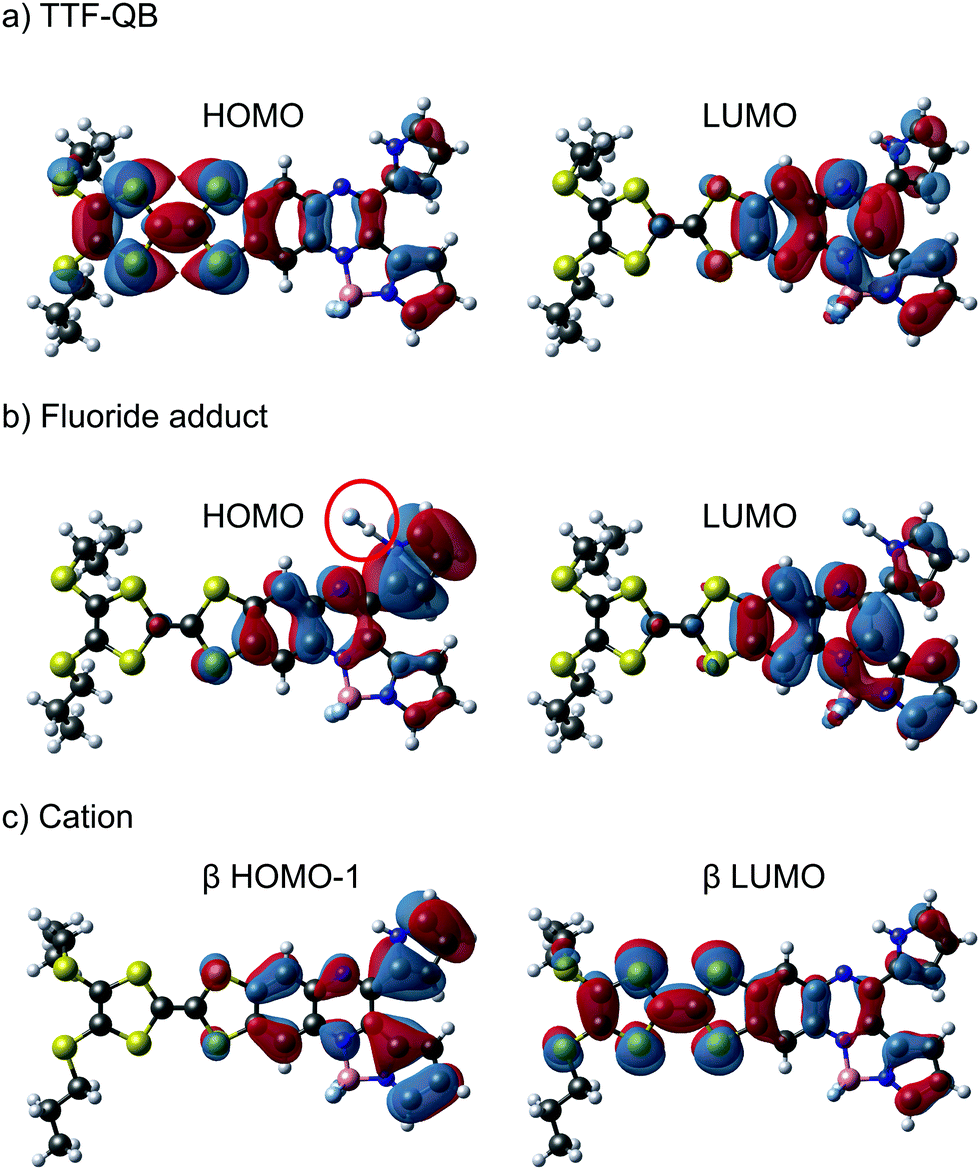 | ||
| Fig. 4 Molecular orbitals of TTF–QB that are involved in the ICT transition, in the neutral state (a), in the presence of fluoride (b) and in the oxidised state (c). The red circle in (b) points to the H-bonded fluoride. | ||
In conclusion, the key finding is that, through external stimuli like fluoride binding or oxidation reaction, the directionality of a photoinduced charge flow within a multichromophoric D–A system can be regulated. TD-DFT calculations allow the characterisation of the ground- and excited electronic states, which in turn reveals the different directions and locations of charge flow within the intricate but still compact molecule. These obtained results pave the way to manipulate photoinduced CT through various pathways by judicious design and chemical modification for potential applications in optoelectronic devices.
This work was financially supported by the Swiss NSF through the NCCR MUST “Molecular Ultrafast Science and Technology”, the Swiss National Foundation (200020_188468), and SNF professorship grants (PP00P2_157615 and PP00P2_187185).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Benniston, Chem. Soc. Rev., 2004, 33, 573 Search PubMed; R. Kaur, F. Possanza, F. Limosani, S. Bauroth, R. Zanoni, T. Clark, G. Arrigoni, P. Tagliatesta and D. M. Guldi, J. Am. Chem. Soc., 2020, 142, 7898 CrossRef CAS; A. Felouat, A. D'Aleo, A. Charaf-Eddin, D. Jacquemin, B. Le Guennic, E. Kim, K. J. Lee, J. H. Woo, J.-C. Ribierre, J. W. Wu and F. Fages, J. Phys. Chem. A, 2015, 119, 6283 CrossRef; A. T. Turley, A. Danos, A. Prlj, A. P. Monkman, B. F. E. Curchod, P. R. McGonigal and M. K. Etherington, Chem. Sci., 2020, 11, 6990 RSC; M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef; J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863 RSC.
- J. E. Klare, G. S. Tulevski, K. Sugo, A. De Picciotto, K. A. White and C. Nuckolls, J. Am. Chem. Soc., 2003, 125, 6030 CrossRef CAS; A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39, 2695 RSC.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736 CrossRef CAS; W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518 CrossRef; O. Inganas, Adv. Mater., 2018, 30, e1800388 CrossRef; S. Ullbrich, J. Benduhn, X. Jia, V. C. Nikolis, K. Tvingstedt, F. Piersimoni, S. Roland, Y. Liu, J. Wu, A. Fischer, D. Neher, S. Reineke, D. Spoltore and K. Vandewal, Nat. Mater., 2019, 18, 459 CrossRef; Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122 CrossRef; C. Huang, S. Chen, K. B. Ornso, D. Reber, M. Baghernejad, Y. Fu, T. Wandlowski, S. Decurtins, W. Hong, K. S. Thygesen and S.-X. Liu, Angew. Chem., Int. Ed., 2015, 54, 14304 CrossRef; A. Amacher, C. Yi, J. Yang, M. P. Bircher, Y. Fu, M. Cascella, M. Graetzel, S. Decurtins and S.-X. Liu, Chem. Commun., 2014, 50, 6540 RSC.
- S. Grunder, R. Huber, V. Horhoiu, M. T. González, C. Schönenberger, M. Calame and M. Mayor, J. Org. Chem., 2007, 72, 8337 CrossRef CAS; X. Liu, X. Li, S. Sangtarash, H. Sadeghi, S. Decurtins, R. Haner, W. Hong, C. J. Lambert and S. X. Liu, Nanoscale, 2018, 10, 18131 RSC; J. Chen and O. S. Wenger, Chem. Sci., 2015, 6, 3582 RSC; H. Pan, G.-L. Fu, Y.-H. Zhao and C.-H. Zhao, Org. Lett., 2011, 13, 4830 CrossRef.
- J. Wu, N. Dupont, S.-X. Liu, A. Neels, A. Hauser and S. Decurtins, Chem. – Asian J., 2009, 4, 392 CrossRef CAS.
- Z. Li, H. Li, S. Chen, T. Froehlich, C. Yi, C. Schoenenberger, M. Calame, S. Decurtins, S.-X. Liu and E. Borguet, J. Am. Chem. Soc., 2014, 136, 8867 CrossRef CAS.
- Y. Chai, X. Liu, B. Wu, L. Liu, Z. Wang, Y. Weng and C. Wang, J. Am. Chem. Soc., 2020, 142, 4411 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Chem. Commun., 2006, 1169 RSC.
- M. R. Bryce, Adv. Mater., 1999, 11, 11 CrossRef CAS.
- C. Yu, E. Hao, T. Li, J. Wang, W. Sheng, Y. Wei, X. Mu and L. Jiao, Dalton Trans., 2015, 44, 13897 RSC.
- K. Tsujimoto, R. Ogasawara, T. Nakagawa and H. Fujiwara, Eur. J. Inorg. Chem., 2014, 3960 CrossRef CAS; K. Tsujimoto, R. Ogasawara and H. Fujiwara, Tetrahedron Lett., 2013, 54, 1251 CrossRef; J. Xiong, L. Sun, Y. Liao, G.-N. Li, J.-L. Zuo and X.-Z. You, Tetrahedron Lett., 2011, 52, 6157 CrossRef; K.-L. Huang, N. Bellec, M. Guerro, F. Camerel, T. Roisnel and D. Lorcy, Tetrahedron, 2011, 67, 8740 CrossRef.
- H.-P. Jia, J. C. Forgie, S.-X. Liu, L. Sanguinet, E. Levillain, F. Le Derf, M. Salle, A. Neels, P. J. Skabara and S. Decurtins, Tetrahedron, 2012, 68, 1590 CrossRef CAS.
- C. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804 CrossRef CAS; C. Jia, S. Liu, C. Tanner, C. Leiggener, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. Commun., 2006, 1878 RSC.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603 CrossRef CAS; H.-P. Jia, J. Ding, Y.-F. Ran, S.-X. Liu, C. Blum, I. Petkova, A. Hauser and S. Decurtins, Chem. – Asian J., 2011, 6, 3312 CrossRef; J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05736c |
| This journal is © The Royal Society of Chemistry 2020 |