
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity prediction in aza-Michael additions without transition state calculations: the Ames test for mutagenicity†
Piers A.
Townsend
 ab and
Matthew N.
Grayson
*b
ab and
Matthew N.
Grayson
*b
aCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath, BA2 7AY, UK
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: M.N.Grayson@bath.ac.uk
First published on 8th October 2020
Abstract
Animal testing remains a contentious ethical issue in predictive toxicology. Thus, a fast, versatile, low-cost quantum chemical model is presented for predicting the risk of Ames mutagenicity in a series of 1,4 Michael acceptor type compounds. This framework eliminates the need for transition state calculations, and uses an intermediate structure to probe the reactivity of aza-Michael acceptors. This model can be used in a variety of settings e.g., the design of targeted covalent inhibitors and polyketide biosyntheses.
With increasing scientific, ethical, and societal drivers to reduce animal testing, the development of novel predictive in silico approaches for chemical safety assessment is vital. In silico methods are low-cost, and are amongst the fastest, most dynamic approaches for the initial safety assessment of chemicals.1 For these reasons, there is great interest in adopting computational approaches in toxicological regulation and legislation.2 Cancer, or carcinogenicity, continues to be a toxicological endpoint of great concern, with over 9 million people dying every year from cancer related illness.3 Mutagenicity is a well-studied endpoint to predict carcinogenicity and is a process by which inheritable cancer-causing genetic changes are induced.4 When DNA mutations arise, the first step often involves the formation of a covalent adduct between a DNA nucleobase and an electrophilic exogenous chemical agent.4,5 The Ames test is a widely used in vitro bioassay for identifying carcinogens and has mutagenicity as the observed endpoint.6 In drug discovery, a positive Ames test result on novel drug candidates can be a significant roadblock in their development; it is indicative of a hazardous chemotype and can often halt clinical trials on potential therapeutics.7 Thus, the in silico prediction of Ames test results provides a number of advantages; a rapid, low-cost assessment at the earliest possible stage in development will save time and money, along with circumnavigating the need for animal testing. Transition state modeling (TSM) has been used extensively for studying chemical reaction mechanisms.8,9 The chemical reactivity of 1,4 Michael Acceptors (MAs) in aza-Michael additions underpins many chemical processes and this reactivity can be assessed with TSM approaches.10 Examples include DNA adduct formation and the design of novel candidates for targeted covalent inhibitors (TCIs).11 TCIs are rationally designed to first bind with their target protein through intermolecular interactions, followed by an important covalent bond forming reaction between the TCI and nucleophilic moieties e.g., amine side chains. The covalent modification leads to increased binding affinities and thus, increased potency. Further, aza-Michael additions are common in ribosomal and non-ribosomal peptide biosynthesis, and have recently been adopted as a strategy in macrolactam polyketide biosynthesis.12
In 2019, we published a model that utilised DFT TSM to predict the Ames test results of 30 1,4 Michael acceptors, with activation free energy and LUMO energies showing significant correlation with mutagenic risk.13 Our results showed that a higher level of theory in the molecular geometry optimisation procedure had a direct effect on the quality of the reactivity predictions made on the original dataset of 19 MAs.14 Despite significant predictive potential shown by the model, TSM can be costly, exhaustively time consuming and challenging for the non-expert. Thus, examination of the toxicant-target potential energy surface without the use of TSM is beneficial for fast, low cost predictions in applications such as industrial chemical risk assessment and the design of novel TCIs. In the reaction between 1,4 MAs and biological nucleophiles, a high energy intermediate (HEI) enolate exists along the reaction pathway (see Fig. 1).15 Calculations on these high energy intermediate structures, that closely resemble the transition states in line with Hammond's postulate, may be broadly extended to the wide variety of toxicologically relevant covalent phenomena discussed above.16 This study presents a novel framework for the prediction of Ames test results using the HEI formed in the reaction between 1,4 Michael acceptors and nucleophilic moieties such as DNA nucleobases. Further, we directly compare the use of fast, low-cost semi-empirical calculations with slower yet more accurate DFT calculations.
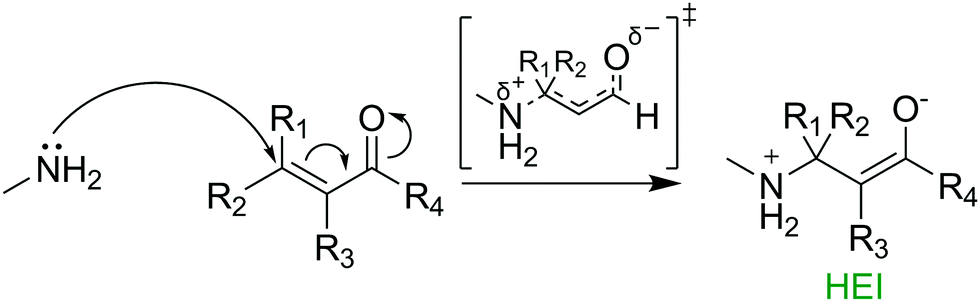 | ||
| Fig. 1 Mechanism for the formation of an enolate intermediate in the reaction between a typical 1,4 Michael acceptor and methylamine. | ||
In this study, 28 1,4 Michael acceptors were selected from our previously published dataset (see ESI,† Fig. S1). Two structures were eliminated from the original dataset. Firstly, 2-bromoacrylic acid was removed due to it being the only carboxylic acid in the group. Secondly, 3-(dichloromethylidene)pyrrolidine-2,5-dione was removed due to an inconclusive Ames test result, leading to uncertainty in the correct classification.17 For the 28 compounds, experimental Ames test results towards the TA100 strain of S. typhimurium (without S9) were taken from two resources: the OECD QSAR Toolbox and a dataset published in 2010 by Cordeiro and co-workers.18,19 Of the 28 compounds, 14 were previously classified as Ames positive, whilst a further 14 were classified as Ames negative. To ensure faster calculation times and reduced computational costs, methylamine was chosen as a surrogate for the most reactive DNA nucleobase, guanine (see Fig. 1 and Fig. S2, ESI†).20 Conformational searches were performed using the MMFF force field, on both the reactants and HEIs of the chosen compounds using Schrödinger's Macromodel (Version 11.3).21 Successfully converged conformations were further optimised in Gaussian 16 (Rev. A.03) using both semi-empirical and DFT methods (see ESI† for full details).22 All semi-empirical optimizations used the Austin Model 1 (AM1) level of theory followed by single point energy calculations at the B3LYP-D3(BJ)/def2-TZVPP-IEFPCM(water) level of theory. DFT structures were calculated at the B3LYP-D3(BJ)/def2-TZVPP-IEFPCM(water) level of theory. These methods were chosen due to extensive use in previous studies of organic chemical reaction mechanisms.23–25
Our previously published model utilised activation energies and molecular orbital energies to predict the Ames test result of 1,4 Michael acceptors.13 Despite the significant predictive potential shown by the model, TSs can be time-consuming and challenging to locate computationally. Further, conformational flexibility can lead to hundreds of individual TSs, ensuring high computational costs. The HEI structure is a minimum on the potential energy surface, as opposed to a saddle point, and thus involves a much simpler calculation procedure than that required to locate TSs (see Fig. S3, ESI†). This demands the question: can the energy difference between the HEI and the reactants (ΔGAM1/ΔGDFT) be used as a quantum chemical descriptor for Ames test predictions (see Fig. S4, ESI†)? Throughout this work, energies of the reactants and high energy intermediates were calculated. ΔGAM1/DFT is computed as follows:
| ΔGAM1/DFT = GHEI − (GMA + GN) | (1) |
Austin Model 1 Semi-empirical calculations have an accuracy known to lie between molecular mechanics (MM) and QM approaches such as DFT and Hartree–Fock.26 Despite the decrease in accuracy compared to higher level methods, semi-empirical calculations are much faster than DFT methods, and thus, show desirable characteristics for the prediction of toxicological phenomena, such as Ames mutagenicity.27 In total, the HEI for 26 of the 28 compounds were successfully optimised using AM1, and the HEI energy difference (ΔGAM1) showed wide-ranging values from 13.8 to 53.1 kcal mol−1 (see Table 1). Ames positive compounds ranged from 13.8 to 37.4 kcal mol−1 whilst Ames negative compounds ranged from 29.3 to 53.1 kcal mol−1. The average ΔGAM1 for Ames positive compounds was 26.1 kcal mol−1 whilst a higher average of 36.2 kcal mol−1 was seen in Ames negative compounds. It is clear that the average energy difference of 10.1 kcal mol−1 between Ames positive and Ames negative compounds is significant. Of the 26 compounds calculated, only two false positives were apparent; two Ames negative compounds (1 and 8, see below for further details) sat within the expected ΔGAM1 region for Ames positive compounds (see Table 1). A single Ames positive compound (14) fell within the expected ΔGAM1 range for Ames negative compounds. Thus, only a single false negative prediction was observed with the AM1 method. Upon inspecting the optimized structures of compounds 3 and 9, it was apparent that a stable HEI could not be isolated at the AM1 level of theory. The optimization results consistently resulted in nucleophile–electrophile distances greater than 5 Å. A number of attempts were made to obtain these intermediates, including geometry optimization with constraints at the C–N atomic coordinates. Once the constrained structure had been optimised to a minimum, the constraint was removed, and a further optimization was performed. The resulting intermediates could not be readily found on the AM1 potential energy surface. Despite these interesting results, for a highly inexpensive computational approach with calculation times much shorter than DFT, often sitting below one minute for AM1 optimizations, a clear trend of separation in ΔGAM1 exists at the semi-empirical AM1 level of theory following DFT SPE correction. Many commonly used (Q)SAR chemical descriptors show greater variation in their values when included in published models.28,29 Thus, the HEI energy difference ΔGAM1 shows great promise for use in multivariate (Q)SAR models targeted towards 1,4 MA toxicity prediction.
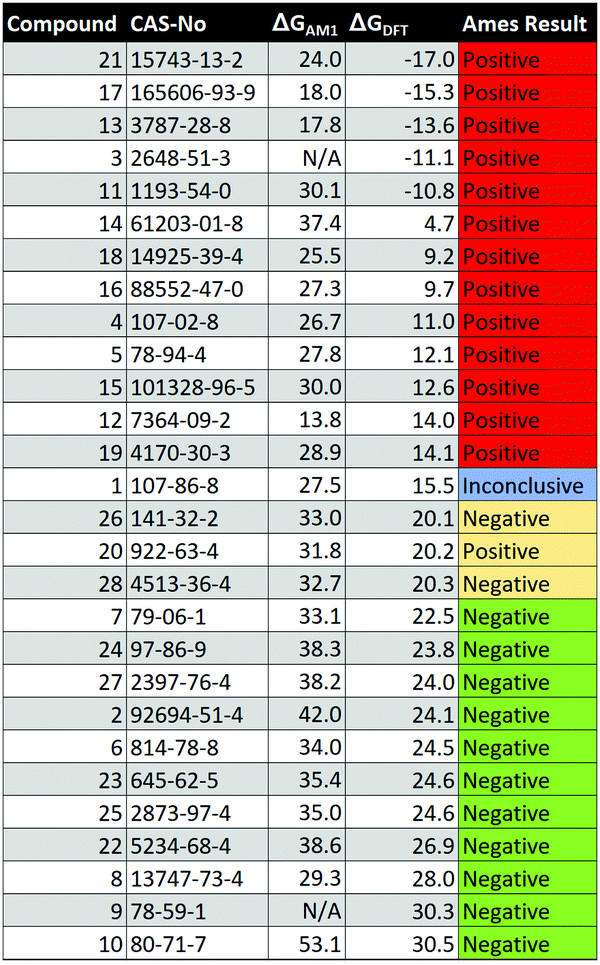
|
Density Functional Theory (DFT) Compared to semi-empirical methods such as AM1, DFT is known to be slower yet more accurate for energies and molecular geometries.30 For 23 of 28 compounds in the dataset, HEIs were successfully found and optimised at the B3LYP-D3(BJ)/def2tzvpp level of theory within the IEFPCM model (water). Compared to AM1, a narrower range of 4.7–30.5 kcal mol−1 was found for the HEI energy difference ΔGDFT. The Ames positive compounds showed ΔGDFT values ranging from 4.7–20.2 kcal mol−1, whilst Ames negative compounds ranged from 20.1–30.5 kcal mol−1. The average ΔGDFT for Ames positive and Ames negative compounds was 12.0 and 24.1 kcal mol−1 respectively, leading to an average energy difference of 12.1 kcal mol−1 between mutagenic and non-mutagenic compounds. For five compounds (3, 11, 13, 17, 21), the HEI could not be found using DFT. However, significant results were observed for each of these structures. There were two similarities between each of the five failed compounds: (i) all compounds have experimentally determined Ames positive results and (ii) each compound had at least one chlorine atom at the β-carbon. For each of the five compounds, the optimised structures were all verified minima on the potential energy surface. However, as opposed to the HEI, a product-type state was observed for each compound, along with the elimination of hydrogen chloride (see Fig. S5, ESI†). The free energy of these product states can be termed GPS. When ΔGDFT was computed for the five failed compounds, GHEI was replaced with GPS and as a result, negative values for ΔGDFT were obtained (see Table 1). These results can be explained by the role that chlorine plays as a leaving group; weak bases are known to be effective leaving groups.31 Further, there is evidence that chlorine containing compounds are often positively classified in the Ames test. The published dataset from Kazius and Bursi highlights a number of chlorine containing toxicophores and their associated mutagenic risk: 15 of 18 α-chlorothioalkanes, 22 of 26 chloroalkenes, 10 of 10 polyhaloalkenes, 18 of 19 1-chloroethyl and 29 of 32 β-haloethoxys were experimentally classified as Ames positive.32 The product states observed in the chlorine containing 1,4 MAs are indicative of stable adduct formation upon exposure to methylamine and thus, nitrogen containing nucleobases such as guanine. From a practical perspective, it is worth noting that no further effort was required to obtain GPS and it is simply the outcome of geometry optimisation and subsequent data analysis. The HEI energy difference calculated for compound 1 led to interest in its mutagenic risk. From our dataset, compound 1 was the only experimentally classified Ames negative compound showing ΔGDFT < 20.0 kcal mol−1, with its ΔGDFT value sitting at 15.5 kcal mol−1. This result led to re-examination of the toxicological literature for this structure. The OECD have previously reported “a failure to product coherent results” for compound 1.33 Although Ames negative results have previously been reported for compound 1, mutagenic activity and Ames positive results have also been reported, leading to uncertainty in the mutagenic risk for this chemical. These results likely explain the magnitude of ΔGDFT for compound 1; by placing ΔGDFT into the Ames positive region, this method successfully captures the mutagenic potential shown by this compound in its OECD assessment. Further, although its ΔGDFT lies in the Ames positive region, it has the highest ΔGDFT value of all structures in the positive region, thereby placing it closer to the negative region than other positive compounds. This may account for the uncertainty in its experimentally derived Ames test result. The energies of ΔGDFT obtained show excellent ability to categorise the Ames test result in 1,4 MAs. There is an area of uncertainty (AOU) at 20 kcal mol−1, with one Ames positive compound (20) at 20.2 kcal mol−1 and a one Ames negative compound (26) at 20.1 kcal mol−1. Benigni et al, however, previously showed that compound 20 exhibits a relatively low molar mutagenicity when compared to other values in their dataset.34 The model constructed in this work shows a divide between ΔGDFT values calculated for Ames positive (mutagenic) and Ames negative (non-mutagenic) compounds. The AOU at 20 kcal mol−1 does exist, and thus, compounds in this region should be treated with caution. Structural features of AP and AN compounds are highlighted and discussed (see ESI,† Fig. S7). To improve the usability of this model, and to utilise this method for the prediction of reactivity in 1,4 MAs, we have proposed a formal protocol (see Fig. 2 and ESI†). To ensure our model was consistent across methods, we performed three sets of additional calculations on the AOU in which either the DFT functional, basis set or solvent model was changed. The results of these calculations showed that ΔGDFT can be used to predict Ames test results at multiple levels of theory (see ESI,† Fig. S8). Further, to test our DFT model, calculations were performed on two common Michael acceptor type pharmaceuticals: warfarin and embelin (minor truncation, see ESI†). ΔGDFT values of 34.3 and 29.6 kcal mol−1 were respectively obtained, agreeing with experiment, and placing them in the Ames negative region.35
 | ||
| Fig. 2 Flow diagram demonstrating the suggested workflow for predicting the Ames test result of a 1,4 MA. | ||
This work has presented a rapid, low-cost procedure for assessing the mutagenic risk of 1,4 Michael acceptor type compounds. In the reaction between 1,4 MAs and methylamine, a high energy intermediate is present on the reaction pathway following the C–N bond forming TS. The use of intermediate structures in place of transition state structures allows for faster, low-cost data acquisition. From a practical standpoint, intermediate structures (minima) are much simpler to compute than TSs (saddle points). The high energy intermediate energy differences (ΔGAM1/ΔGDFT) were calculated and compared at two levels of theory: the semi-empirical method AM1 and DFT. It was shown that AM1 provides a suitable, low-cost framework for categorising Ames test results. However, false negative predictions did occur, and a few compounds could not be optimised at the AM1 level of theory. DFT results showed excellent performance in categorising Ames test results based on the calculated value of ΔGDFT. It can be deduced that compounds with ΔGDFT < 19.0 kcal mol−1 are likely to be Ames positive, whilst compounds with ΔGDFT > 21.0 kcal mol−1 are likely to be Ames negative. This method can be used in the preliminary safety assessment of new and pre-existing pharmaceutically relevant 1,4 MAs. Further, the model presented provides a powerful template to probe aza-Michael addition reactivity profiles in a variety of settings e.g., the design of targeted covalent inhibitors and polyketide biosyntheses. Both settings often utilise MAs, and may benefit from a method that directly predicts and quantifies the reactivity of different MAs towards nitrogen containing nucleophiles, without the need for TS calculations. This model allows the steric and energetic details of aza-Michael additions to be studied and elucidated using an approach that directly considers a HEI in place of a TS.
This work was supported by the Engineering and Physical Sciences Research Council (EP/L016354/1) and the University of Bath. This work was completed using the Balena HPC service at the University of Bath.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Hasselgren, et al. , Regul. Toxicol. Pharmacol., 2019, 107, 104403 CrossRef CAS.
- N. Gellatly and F. Sewell, Comput. Toxicol., 2019, 11, 82–89 CrossRef.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef.
- S. J. Enoch, C. M. Ellison, T. W. Schultz and M. T. D. Cronin, Crit. Rev. Toxicol., 2011, 41, 783–802 CrossRef CAS.
- E. Eder, C. Hoffman, H. Bastian, C. Deininger and S. Scheckenbach, Environ. Health Perspect., 1990, 88, 99–106 CAS.
- B. Ames, in Chemical Mutagens: Principles and Methods for their Detection, 1971.
- P. Mccarren, C. Springer and L. Whitehead, Cheminf, 2011, 3, 51 CrossRef CAS.
- D. M. Sedgwick, M. N. Grayson, S. Fustero and P. Barrio, Synth., 2018, 50, 1935–1957 CrossRef CAS.
- E. H. Krenske, R. C. Petter, Z. Zhu and K. N. Houk, J. Org. Chem., 2011, 76, 5074–5081 CrossRef CAS.
- P. Townsend and M. Grayson, Chem. Res. Toxicol. DOI:10.1021/acs.chemrestox.0c00113.
- M. H. Johansson, Mini Rev. Med. Chem., 2012, 12, 1330–1344 CAS.
- A. Miyanaga, Nat. Prod. Rep., 2019, 36, 531–547 RSC.
- P. A. Townsend and M. N. Grayson, J. Chem. Inf. Model., 2019, 59, 5099–5103 CrossRef CAS.
- T. E. H. Allen, M. N. Grayson, J. M. Goodman, S. Gutsell and P. J. Russell, J. Chem. Inf. Model., 2018, 58, 1266–1271 CrossRef CAS.
- S. J. Enoch and D. W. Roberts, Chem. Res. Toxicol., 2013, 26, 767–774 Search PubMed.
- N. Agmon, J. Chem. Soc., 1978, 74, 388–404 Search PubMed.
- B. A. Freeman, R. E. Wilson, R. G. Binder and W. F. Haddon, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2001, 490, 89–98 CrossRef CAS.
- A. Pérez-Garrido, A. M. Helguera, F. G. Rodríguez and M. N. D. S. Cordeiro, Dent. Mater., 2010, 26, 397–415 CrossRef.
- OECD, OECD QSAR Toolbox, http://www.oecd.org/chemicalsafety (Acc. August 2020).
- D. Gillingham, S. Geigle and O. Anatole Von Lilienfeld, Chem. Soc. Rev., 2016, 45, 2637–2655 RSC.
- Macromodel, Schrödinger, New York, NY, 2019 Search PubMed.
- M. J. Frisch, et al., Gaussian 16 (Rev A.03), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- M. N. Grayson, J. Org. Chem., 2017, 82, 4396–4401 CrossRef CAS.
- R. Rittner, L. C. Ducati, C. F. Tormena, B. C. Fiorin and C. B. Braga, Spectrochim. Acta, 2011, 79, 1071–1076 CrossRef CAS.
- S. S. Stoyanov, D. Y. Yancheva and B. A. Stamboliyska, Comput. Theor. Chem., 2014, 1046, 57–63 CrossRef CAS.
- W. Thiel, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 145–157 CAS.
- J. P. McNamara and I. H. Hillier, Phys. Chem. Chem. Phys., 2007, 9, 2362–2370 RSC.
- F. Gramatica, P. Vighi and M. Consolaro, Chemosphere, 2001, 42, 873–883 CrossRef.
- R. S. Hunter and G. D. Sinks, SAR QSAR Environ. Res., 1995, 3, 27–36 CrossRef.
- L. Simón and J. M. Goodman, Org. Biomol. Chem., 2011, 9, 689–700 RSC.
- G. Patlewicz, A. O. Aptula, E. Uriarte, D. W. Roberts, P. S. Kern, G. F. Gerberick, I. Kimber, R. J. Dearman, C. A. Ryan and D. A. Basketter, SAR QSAR Environ. Res., 2007, 18, 515–541 CrossRef CAS.
- J. Kazius, R. McGuire and R. Bursi, J. Med. Chem., 2005, 48, 312–320 CrossRef CAS.
- OECD, SIDS Assessment Report – 3-Methyl-2-Butenal, Arona, Italy, 2003.
- R. Benigni, L. Conti, R. Crebelli, A. Rodomonte and M. R. Vari, Environ. Mol. Mutagen., 2005, 46, 268–280 CrossRef CAS.
- K. R. Sumalatha and M. Sreepriya, Int. J. Pharma Bio Sci., 2015, 6, P290–P296 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05681b |
| This journal is © The Royal Society of Chemistry 2020 |