
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMmfL catalyses formation of a phosphorylated butenolide intermediate in methylenomycin furan biosynthesis†
Shanshan
Zhou
 a,
Nicolas R.
Malet
a,
Lijiang
Song
a,
Christophe
Corre
abc and
Gregory L.
Challis
*acd
a,
Nicolas R.
Malet
a,
Lijiang
Song
a,
Christophe
Corre
abc and
Gregory L.
Challis
*acd
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: g.l.challis@warwick.ac.uk
bSchool of Life Sciences, University of Warwick, Coventry, CV4 7AL, UK
cWarwick Integrative Synthetic Biology Centre, University of Warwick, Coventry, CV4 7AL, UK
dDepartment of Biochemistry and Molecular Biology, Centre of Excellence for Innovations in Peptide and Protein Science, Monash University, Clayton, VIC 3800, Australia
First published on 22nd October 2020
Abstract
Using a combination of a synthetic substrate analogue and product standard, MmfL, a homologue of the γ-butyrolactone biosynthetic enzyme AfsA, was shown to catalyse the condensation of dihydroxyacetone phosphate with a β-ketoacyl thioester to form a phosphorylated butenolide intermediate in the biosynthesis of the methylenomycin furans, which induce methlenomycin antibiotic production in Streptomyces coelicolor A3(2). AfsA homologues are also involved in the biosynthesis of 2-akyl-4-hydroxy-3-methyl butenolide inducers of antibiotic production in other Streptomyces species, indicating that diverse signalling molecules are assembled from analogous phosphorylated butenolide intermediates.
Bacterial specialized metabolites are one of the largest reservoirs of bioactive natural products, including antimicrobials, antitumor agents, immunosuppressants, anthelmintics and insecticides, herbicides and antifungal agents.1 The biosynthesis of bacterial specialized metabolites is often regulated by signalling molecules known as autoregulators, autoinducers or hormones.2,3 These diffusible small molecules induce specialized metabolite production, and in some cases morphogenesis, by binding to specific transcriptional regulators.
Actinobacteria are prolific producers of specialized metabolites that employ various signalling molecules to control their biosynthesis, including γ-butyrolactones (GBLs), 2-alkyl-4-hydroxymethylfuran-3-carboxylic acids (AHFCAs),3,4 2-alkyl-4-hydroxy-3-methylbutenolides (AHMBs) and avenolide (Fig. 1a–d).5–7 GBLs have been identified in a range of Streptomyces species, such as virginiae butanolides (VBs) A–E from Streptomyces virginiae, IM-2 from Streptomyces lavendulae FRI-5, SCBs 1–8 from Streptomyces coelicolor A3(2) and A-factor from Streptomyces griseus (Fig. 1a and e).8–13 These molecules all have similar structures, differing only in the oxidation state, hydroxyl group configuration, position of methyl branches and length of the alkyl chain.
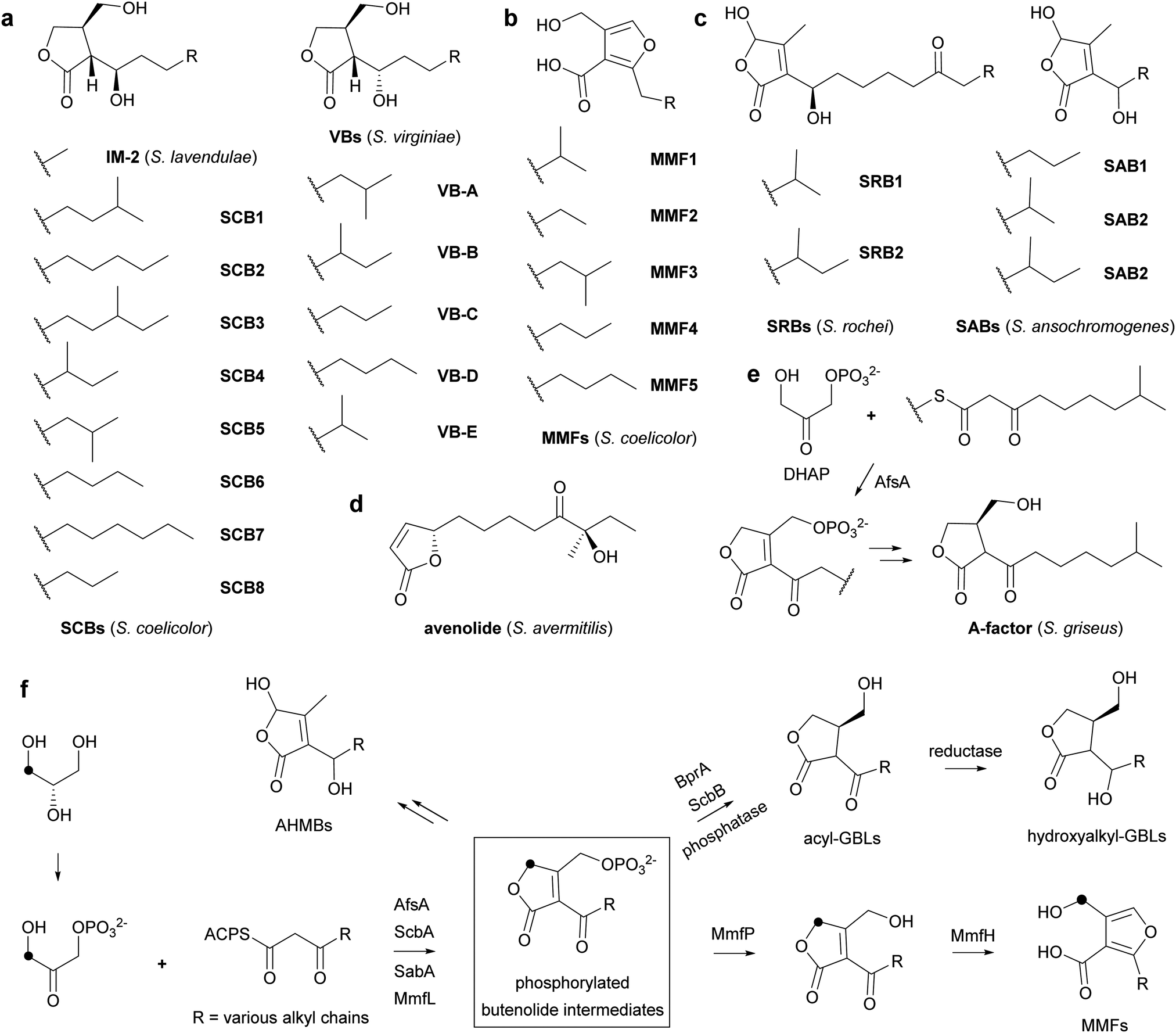 | ||
| Fig. 1 Structures of Streptomyces signalling molecules and proposed pathways for their biosynthesis. (a) GBLs, (b) AHFCAs, (c) AHMBs, (d) avenolide. (e) AfsA catalyses the condensation of DHAP with a β-ketothioester in A-factor biosynthesis. (f) Proposed pathways for the biosynthesis of GBLs, AHMBs and AHFCAs, involving intermediates with a common phosphorylated butenolide core structure (boxed). BprA catalyses reduction of the phosphorylated butenolide intermediate in A-factor biosynthesis to the corresponding butanolide. ScbB is a BprA orthologue that is proposed to catalyse an analogous reaction in SCB biosynthesis. Additional reductases convert the keto group to differently configured hydroxyl groups in the biosynthesis of VBs and SCBs/IM-2. Black dots indicate the site of incorporation of the 13C-label from [3-13C]-L-glycerol into DHAP, the butenolide-containing intermediates and the MMFs. | ||
A-factor, the first GBL to be discovered, induces production of the antibiotic streptomycin and the yellow pigment grixazone in S. griseus, in addition to morphological differentiation.13,14 AfsA has been identified as key enzyme in A-factor biosynthesis that is proposed to condense an acyl carrier protein (ACP)-bound β-keto thioester intermediate in fatty acid biosynthesis with a 3-carbon glycolytic intermediate. It has been reported to catalyse the condensation of the N-acetylcysteamine (NAC) thioester of 8-methyl-3-oxononanoate with dihydroxyacetone phosphate (DHAP) resulting in the formation of a phosphorylated butenolide (Fig. 1e).15 Reduction of this butenolide to the corresponding butanolide by BprA, followed by dephosphorylation yields A-factor.
AfsA homologues have been reported to be involved in the biosynthesis of other GBLs. For example, ScbA participates in the assembly of SCBs in S. coelicolor and has been proposed to consist of two divergent domains with similarity to the fatty acid dehydratases FabA and FabZ.16,17 Interestingly, they are also involved in the biosynthesis of other classes of Actinobacterial signalling molecules, such as AHFCAs and AHMBs. Thus, MmfL and SabA participate in the assembly of methylenomycin furans (MMFs) 1–5 and SABs 1–3, AHFCAs and AHMBs that induce the production of methylenomycin and nikkomycin antibiotics in S. coelicolor and Streptomyces ansochromogenes, respectively (Fig. 1b and c and Fig. S1, ESI†).4,7 We hypothesize that these enzymes catalyse analogous reactions to that catalysed by AfsA and that GBLs, AHFCAs and AHMBs are all assembled via intermediates with a common phosphorylated butenolide core structure (Fig. 1f). Incorporation experiments with stereospecifically 13C-labelled glycerols,18 which show that the hydroxymethyl group of the MMFs derives from the corresponding functional group in DHAP, support this proposal (Fig. 1f). However, there is currently no direct evidence for the role played by MmfL in MMF biosynthesis. Here, we report enzymatic characterization of purified recombinant MmfL using a chemically synthesized substrate analogue and standard of the dephosphorylated product. These experiments establish that MMF biosynthesis proceeds via a phosphorylated butenolide intermediate, which undergoes dephosphorylation and rearrangement to form the AHFCA skeleton.
We overproduced MmfL in E. coli as a soluble N-terminal His6 fusion protein, enabling purification to homogeneity using nickel affinity chromatography (Fig. S2a, ESI†). ESI-Q-TOF-MS confirmed the identity of the purified protein (Fig. S2b, ESI†). The substrate scope of MmfL appears to be comparatively broad, as MMFs with various alkyl chains have been isolated from S. coelicolor.4 We synthesised the NAC thioester of 3-ketooctanoate 1 (ESI†),19 which mimics the presumed β-ketoacyl-ACP precursor of MMF5, and used this to investigate the enzymatic activity of MmfL (Fig. 2a). His6-MmfL (15 μM), was incubated with 1 (400 μM) and DHAP (400 μM) at 25 °C for 90 min and the mixture was treated with shrimp alkaline phosphatase (SAP) to dephosphorylate the anticipated product of the reaction, phosphorylated butenolide 2. A similar strategy was used to overcome problems with detection of the phosphorylated butenolide product in LC-MS analyses of AfsA-catalysed reactions.15 A species with m/z = 235.0936, corresponding to [M + Na]+ for butenolide 3 (calculated m/z for C11H1604Na+: 235.0941) was readily detected in UHPLC-ESI-Q-TOF-MS analyses of the phosphatase-treated reaction mixture (Fig. 2b). This species was absent from control reactions in which either DHAP or 1 was omitted, or heat-denatured MmfL was employed, confirming that it results from the MmfL-catalysed condensation of 1 with DHAP.
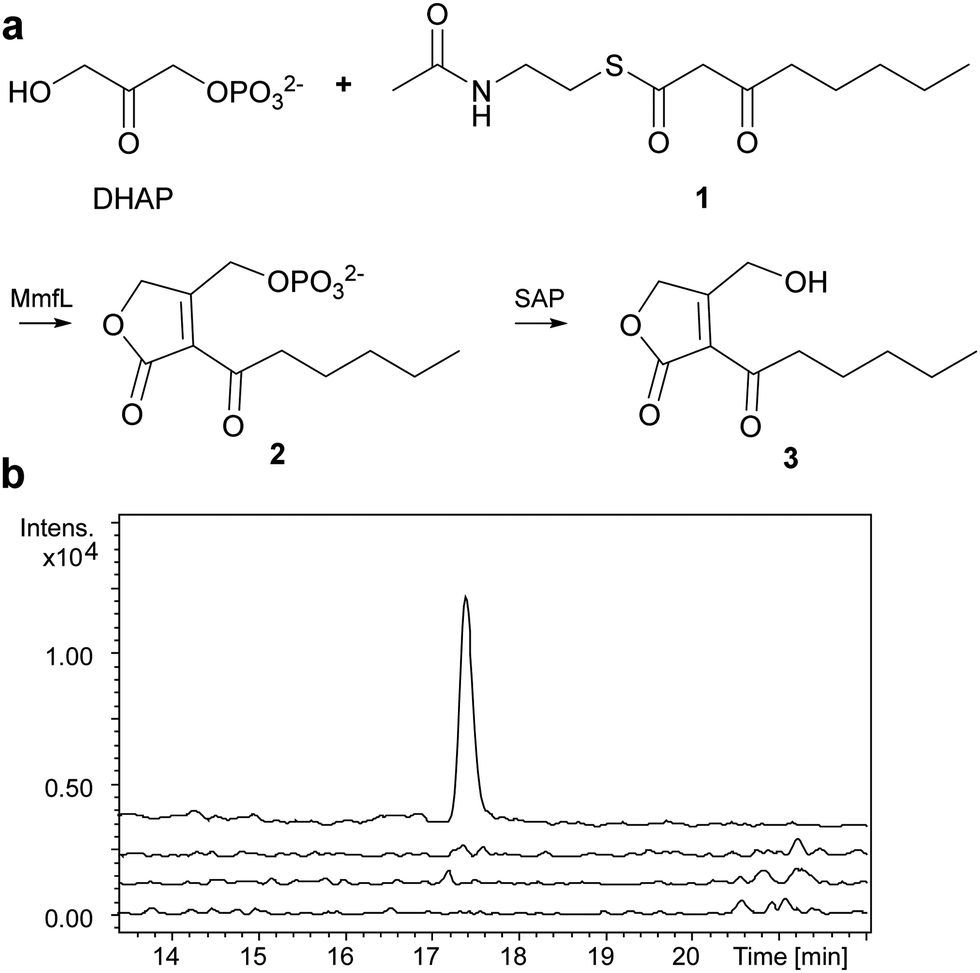 | ||
| Fig. 2 (a) MmfL-catalysed condensation of DHAP with 1. Products were dephosphorylated by treating with shrimp alkaline phosphatase (SAP) prior to analysis. (b) Extracted ion chromatograms (EICs) for m/z = 235.0941, corresponding to [M + Na]+ for 3, from UHPLC-ESI-Q-TOF-MS analyses of the MmfL-catalysed reaction (top trace) and control reactions containing heat-denatured MmfL (second from top), lacking DHAP (second from bottom) and lacking 1 (bottom). | ||
Butenolides such as 3 are reported in the literature to be unstable20 and this proved to be the case for the product of the MmfL-catalysed reaction, which decomposed during attempts to purify it using preparative HPLC. Thus, we synthesised an authentic standard of 3 to compare with the dephosphorylated product of the enzymatic reaction. Using methodology reported by Sello and co-workers, we prepared the tert-butyldimethylsilyl (TBDMS)-protected derivative 4 of butenolide 3 (Fig. 3a).21 The structure of 4, which was also found to be somewhat unstable and degraded during prolonged storage at −20 °C, was confirmed using HRMS in combination with 1H and 13C NMR spectroscopy (ESI†). Removal of the TBDMS group from 4 under mild conditions (HF–pyridine complex, THF, 0 °C for 40 min), followed by immediate UHPLC-ESI-Q-TOF-MS analysis confirmed that it has the same retention time as the dephosphorylated product of the MmfL-catalysed reaction (Fig. 3b). Moreover, identical fragmentation patterns were observed in MS/MS analyses of the [M + H]+ ions corresponding to the products of the two reactions (Fig. 3c). We thus conclude that the product of the MmfL-catalysed condensation of 1 with DHAP is phosphorylated butenolide 2 (Fig. 2).
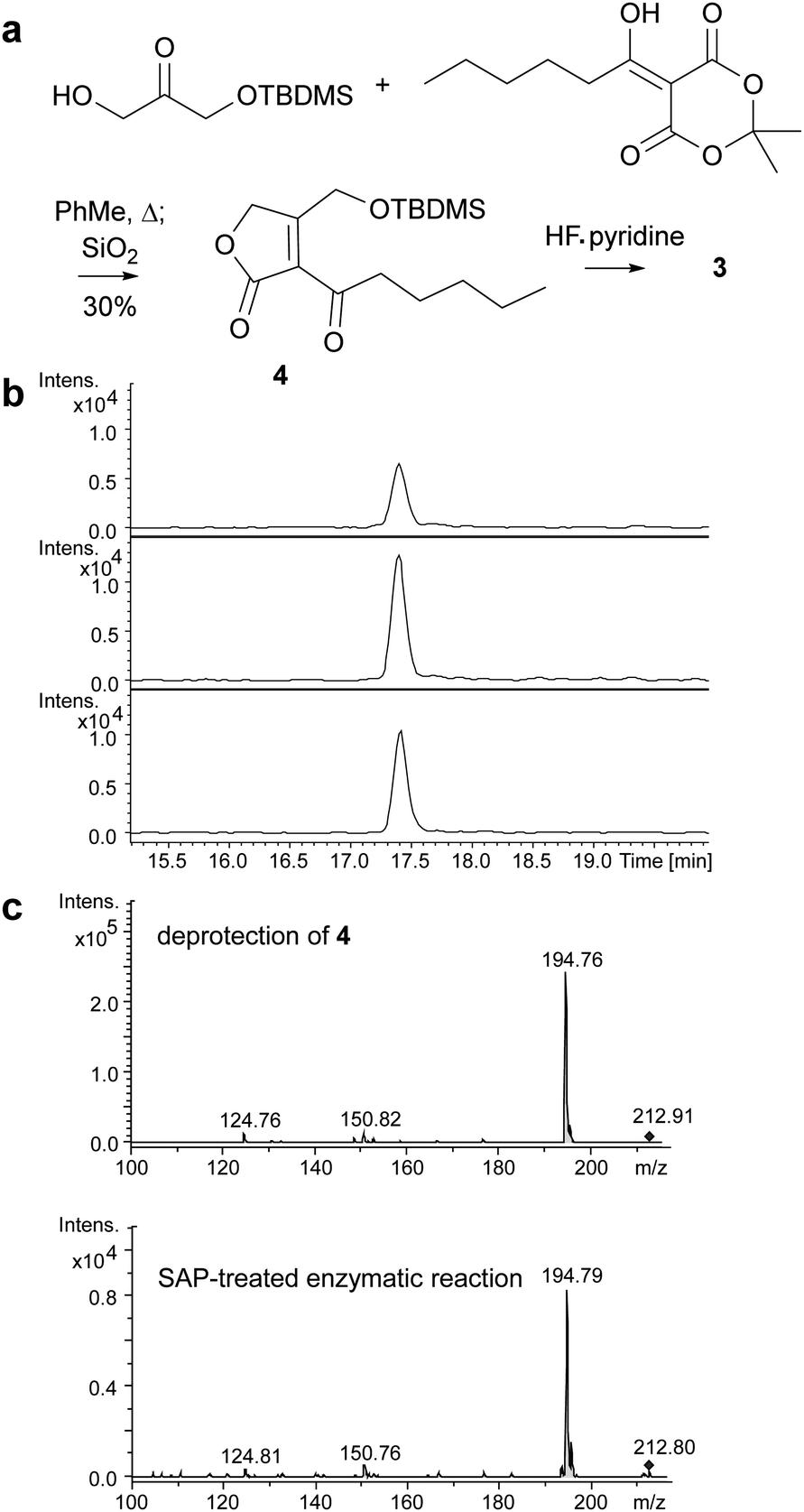 | ||
| Fig. 3 (a) Synthesis of TBDMS-protected butenolide 4. (b) EICs for m/z = 235.0941, corresponding to [M + Na]+ for 3, from UHPLC-ESI-Q-TOF-MS analyses of the dephosphorylated product of the MmfL-catalysed reaction (top), the deprotection of 4 (middle), and a mixture of the products of these two reactions (bottom). (c) MS/MS spectra of the m/z = 213 ion, corresponding to [M + H]+ for 3, observed for the products resulting from the deprotection of 4 (top) and the SAP-treated MmfL-catalysed reaction (bottom). The parent ion is marked with a diamond. | ||
In addition to MmfL, two other enzymes, MmfP and MmfH, are known to be required for MMF biosynthesis in S. coelicolor (Fig. S1a, ESI†).22 MmfP is a putative phosphatase that likely catalyses dephosphorylation of the phosphorylated butenolide intermediates formed by MmfL to form the corresponding 2-acyl-3-hydroxymethylbutenolides (Fig. 1). This suggests that MmfH, which exhibits sequence similarity to Flavin-dependent oxidoreductases, catalyses conversion of the 2-acyl-3-hydroxymethylbutenolides formed by MmfP to the MMFs (Fig. 1). While further experiments are required to substantiate this hypothesis, a plausible mechanism for this transformation involves hydroxylation of C-4 in the butenolide intermediates by Flavin-hydroperoxide, generated by the reaction of MmfH-bound FADH2 with molecular oxygen (Fig. 4). The resulting 2-acyl-3-hydroxymethyl-4-hydroxybutenolides would be expected to undergo facile ring opening, enabling furan formation via a cyclisation–dehydration–tautomerisation cascade (Fig. 4). This yields the corresponding 2-alkyl-4-formyl-furan-3-carboxylic acids, which upon reduction of the formyl group (possibly by MmfH-bound FADH2) would afford the MMFs (Fig. 4). Consistent with this mechanism, 18O is incorporated specifically into the hydroxymethyl group of MMF1 when S. coeliclor is grown under an 18O2 atmosphere (Fig. 4 and Fig. S12 and S13, ESI†).
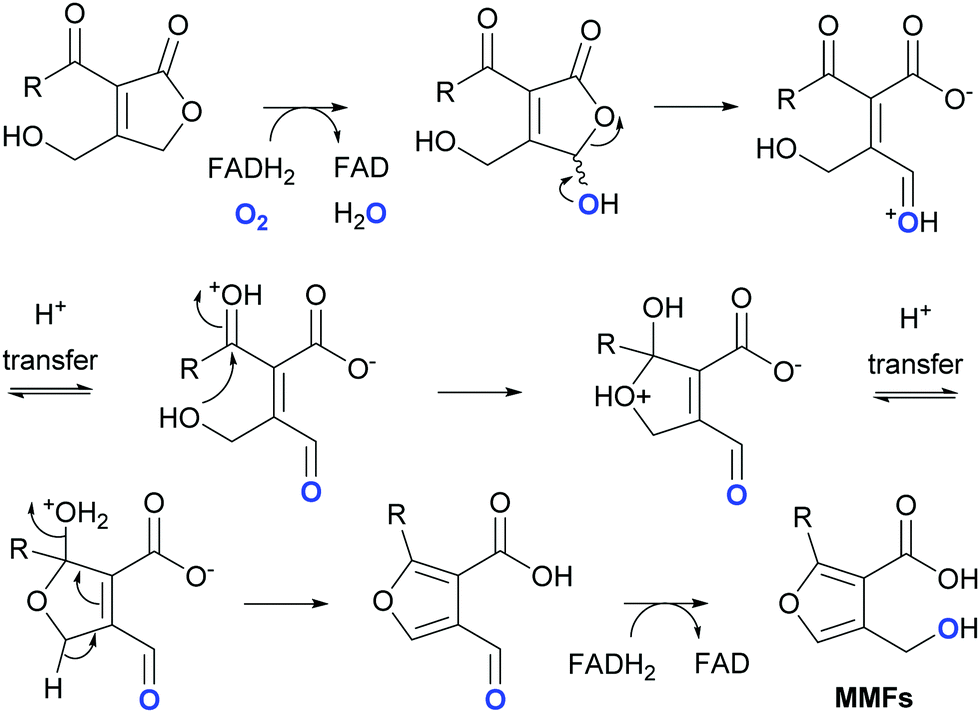 | ||
| Fig. 4 Proposed mechanism for MmfH-catalysed conversion of 2-acyl-3-hydroxymethylbutenolides generated by MmfP to the MMFs. The O atom labelled in the MMFs when S. coelicolor is grown under an 18O2 atmosphere is highlighted in blue. | ||
Very recently, we and others have reported the discovery of gladiostatin (also called gladiofungin A) as the product of a cryptic trans-AT polyketide synthase (PKS) in Burkholderia gladioli.23,24 This unique member of the glutarimide family of antibiotics contains a 2-acyl-4-hydroxy-3-methylbutenolide that is closely related to the butenolide core of the AHMB signalling molecules. We showed that an AfsA-like domain appended to the C-terminus of the PKS releases the fully assembled chain by condensing it with DHAP, resulting in the same type of phosphorylated butenolide intermediate as that produced by AfsA and MmfL in A-factor and MMF biosynthesis. Accordingly, we have proposed “phosphorylated butenolide synthase” as a collective name for this enzyme family.23 It seems likely that the biosynthesis of AHMB signalling molecules such as the SRBs and the SABs, which also involves AfsA homologues, proceeds via analogous phosphorylated butenolide intermediates to those involved in the biosynthesis of GBLs and AHFCAs (Fig. 1f). Together these findings illuminate a fascinating example of how Nature is able to evolve pathways for the assembly of diverse molecular architectures from a common type of biosynthetic intermediate.
This research was supported by the BBSRC (BB/J014532/1; BB/K002341/1 and BB/E008003/1 to G. L. C.; and BB/M022765/1 to C. C.), and the University of Warwick (CIS and WCPRS awards to S. Z. and N. M., respectively). G. L. C. was a recipient of a Wolfson Merit Award from the Royal Society (WM130033).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS.
- A. Camilli and B. L. Bassler, Science, 2006, 311, 1113 CrossRef CAS.
- H. U. van der Heul, B. L. Bilyk, K. J. McDowall, R. F. Seipke and G. P. van Wezel, Nat. Prod. Rep., 2018, 35, 575 RSC.
- C. Corre, L. Song, S. O'Rourke, K. F. Chater and G. L. Challis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17510 CrossRef CAS.
- S. Kitani, K. T. Miyamoto, S. Takamatsu, E. Herawati, H. Iguchi, K. Nishitomi, M. Uchida, T. Nagamitsu, S. Omura, H. Ikeda and T. Nihira, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16410 CrossRef CAS.
- K. Arakawa, N. Tsuda, A. Taniguchi and H. Kinashi, ChemBioChem, 2012, 13, 1447 CrossRef CAS.
- W. Wang, J. Zhang, X. Liu, D. Li, Y. Li, Y. Tian and H. Tan, J. Biol. Chem., 2018, 293, 20029 CrossRef CAS.
- S. Sakuda and Y. Yamada, Tetrahedron Lett., 1991, 32, 1817 CrossRef CAS.
- K. Hashimoto, T. Nihira, S. Sakuda and Y. Yamada, J. Ferment. Bioeng., 1992, 73, 449 CrossRef CAS.
- E. Takano, T. Nihira, Y. Hara, J. J. Jones, C. J. Gershater, Y. Yamada and M. Bibb, J. Biol. Chem., 2000, 275, 11010 CrossRef CAS.
- N.-H. Hsiao, S. Nakayama, M. E. Merlo, M. de Vries, R. Bunet, S. Kitani, T. Nihira and E. Takano, Chem. Biol., 2009, 16, 951 CrossRef CAS.
- J. D. Sidda, V. Poon, L. Song, W. Wang, K. Yang and C. Corre, Org. Biomol. Chem., 2016, 14, 6390 RSC.
- O. Hara and T. Beppu, J. Antibiot., 1982, 35, 349 CrossRef CAS.
- Y. Ohnishi, Y. Furusho, T. Higashi, H. K. Chun, K. Furihata, S. Sakuda and S. Horinouchi, J. Antibiot., 2004, 57, 218 CrossRef CAS.
- J.-y. Kato, N. Funa, H. Watanabe, Y. Ohnishi and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2378 CrossRef CAS.
- J. P. Gomez-Escribano, L. Song, D. J. Fox, V. Yeo, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 2716 RSC.
- N.-H. Hsiao, J. Söding, D. Linke, C. Lange, C. Hertweck, W. Wohlleben and E. Takano, Microbiology, 2007, 153, 1394 CrossRef CAS.
- C. Corre, S. W. Haynes, N. Malet, L. Song and G. L. Challis, Chem. Commun., 2010, 46, 4079 RSC.
- I. H. Gilbert, M. Ginty, J. A. O'Neill, T. J. Simpson, J. Staunton and C. L. Willis, Bioorg. Med. Chem. Lett., 1995, 5, 1587 CrossRef CAS.
- S. Sakuda, S. Tanaka, K. Mizuno, O. Sukcharoen, T. Nihira and Y. Yamada, J. Chem. Soc., Perkin Trans. 1, 1993, 2309 RSC.
- J. B. Morin, K. L. Adams and J. K. Sello, Org. Biomol. Chem., 2012, 10, 1517 RSC.
- S. O’Rourke, A. Wietzorrek, K. Fowler, C. Corre, G. L. Challis and K. F. Chater, Mol. Microbiol., 2009, 71, 763 CrossRef.
- I. T. Nakou, M. Jenner, Y. Dashti, I. Romero-Canelón, J. Masschelein, E. Mahenthiralingam and G. L. Challis, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009007.
- S. P. Niehs, J. Kumpfmüller, B. Dose, R. F. Little, K. Ishida, L. V. Florez, M. Kaltenpoth and C. Hertweck, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202005711.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and small molecule/protein characterisation data. See DOI: 10.1039/d0cc05658h |
| This journal is © The Royal Society of Chemistry 2020 |