
Selective photoredox decarboxylation of α-ketoacids to allylic ketones and 1,4-dicarbonyl compounds dependent on cobaloxime catalysis†
Qing-Xiao
Tong  a
and
Jian-Ji
Zhong
*a
a
and
Jian-Ji
Zhong
*a
a
and
Jian-Ji
Zhong
*a
Abstract
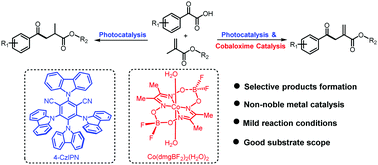
A photoredox/cobaloxime co-catalyzed coupling reaction of α-ketoacids and methacrylates to obtain allylic ketones is described. Without the cobaloxime catalyst, 1,4-dicarbonyl compounds are generated. The cobaloxime catalyst enables dehydrogenation to generate the formation of new olefins. The generality, good substrate scope and mild conditions are good features in the photoredox/cobaloxime catalysis protocol, and this method will provide new opportunities for the functionalization of more olefins.
 Please wait while we load your content...
Please wait while we load your content...

