
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamically tuning transient silicone polymer networks with hydrogen bonding†
Akop
Yepremyan
a,
Andrew
Osamudiamen
a,
Michael A.
Brook
 a and
Andrea
Feinle
*ab
a and
Andrea
Feinle
*ab
aMcMaster University, Department of Chemistry and Chemical Biology, 1280 Main Street West, Hamilton, ON L8S 4M1, Canada
bParis-Lodron University Salzburg, Department of Chemistry and Physics of Materials, Jakob-Haringer Str. 2a, Salzburg 5020, Austria. E-mail: andrea.feinle@sbg.ac.at
First published on 13th October 2020
Abstract
Supramolecular polymers are composed of non-covalently connected chains and characterized by high chain dynamics. The viscoelastic behavior of supramolecular telechelic sugar-siloxanes – ranging from solids to viscous fluids able to form transient polymer networks – is readily tuned by the fraction of internal HO groups that can intermolecularly form hydrogen bonds.
Supramolecular polymers have attracted enormous interest in recent years due to their dynamic nature and exceptional responsiveness to external stimuli. In these systems, transient non-covalent, physical interactions based on metal coordination,1–5 hydrophobic interactions,6–8 π–π stacking,9 protein–protein interactions,10 Lewis-acid Lewis-base interactions,11 or hydrogen bonding12–16 partially or fully replace covalent bonds between polymer chains. The network is non-permanent on experimental timescales because the physical crosslinks are in a permanent equilibrium between their associated and dissociated states; the location of the equilibrium can be influenced by the associative interaction strength, the molecular weight of the polymer, as well as by external parameters such as temperature and the application of shear.12,17 Lewis et al., for example, recently studied the influence of various side groups on poly(butylacrylate) on the intermolecular bonding strength and rheological behavior of the polymer.14 Copolymers bearing weak hydrogen-bonding groups behaved like unentangled melts with no network formation under shear, whereas copolymers with strong hydrogen-bonding groups formed soft and elastic solids.
Given the importance of silicones in many critical applications, such as in medicine (e.g., catheters) or aerospace (e.g., gaskets), we focused on the development of tuneable silicone supramolecular polymers. A few studies have examined the introduction of non-covalent, physical crosslinks into silicone backbones with the aim to improve their recyclability and environmental footprint.18–20 Reports of supramolecular silicone fluids with distinct viscoelastic properties and the ability to transform from the liquid into a rubbery state, however, are rare. Vidal et al. recently reported an approach for the preparation of a dynamically crosslinked polymer network by the combination of Lewis base amine-modified polydimethylsiloxane (PDMS) and Lewis acid organoborane-modified polystyrene (PS).11 The prepared fluids transformed into a solid-like transient polymeric network (TPN) under fast deformation and converted back into its viscous state on long time-scales. We recently reported analogous behavior for linear sugar-modified silicone surfactants (gluconamidosilicones).21 The prepared fluids possess distinct viscoelastic properties and, additionally, excellent damping properties that were attributed to dynamic crosslinks between the gluconamidosilicone chains that allowed the polymer to form a TPN under shear. In this paper, we report the number of hydrogen bonds required on a silicone polymer for a TPN to form, describe the relationship between the hydroxy (HO)/dimethylsiloxane (Me2SiO (D unit)) ratio to the dynamic viscosity and viscoelastic properties of sugaramidosilicones, and show how intermolecular linkages are readily interrupted.
A library of sugaramidosilicones was prepared in which (i) the silicone chain length n was varied from n = 9, 38, 65 to 673, and (ii) the total number x of hydroxy groups attached to the linear aminopropylsilicone chains was increased from x = 4 to 8 and then to 10. Both the silicone chain length and the overall number of hydroxy groups are included in the sample nomenclature: SASn-x represents a sugaramidosilicone with n being the average silicone backbone chain length and x giving the overall number of hydroxy groups per silicone chain. SAS65-8, for example, represents a sugaramidosilicone with 65 dimethylsiloxane repeating units and 4 hydroxy groups on either side of the chain. These sugaramidosilicones were prepared simply and in good yield by opening the respective lactone-derivatives bearing different numbers of hydroxy groups with telechelic aminopropylsilicones (Fig. 1).
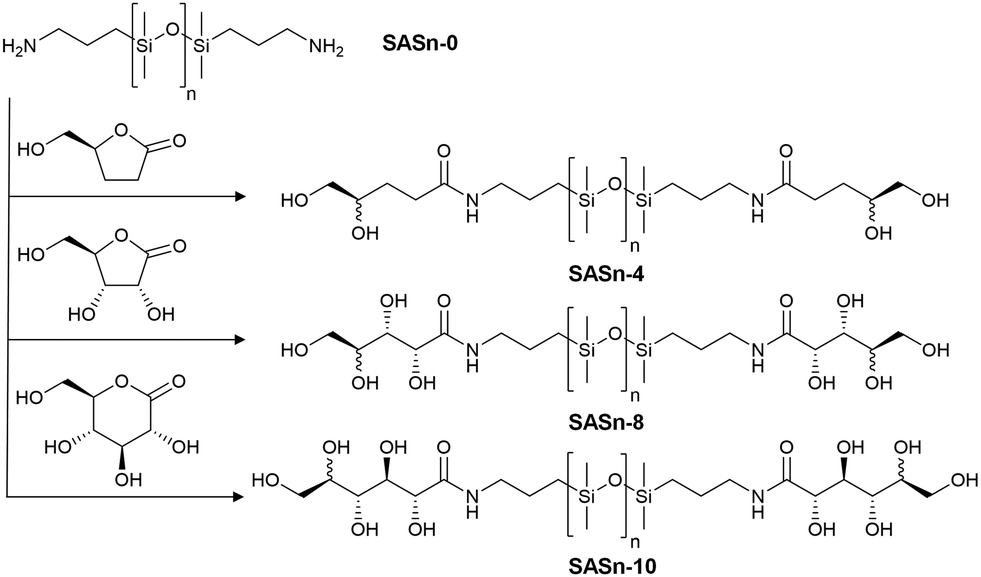 | ||
| Fig. 1 Reaction of aminopropylsilicones (SASn-0) of various silicone chain lengths (n = 9, 38, 65, 673) with different lactone derivatives to give sugaramidosilicones with 4, 8, and 10 hydroxy groups, respectively (SASn-4, SASn-8, and SASn-10). | ||
The dynamic viscosities of the starting materials and sugaramidosilicones were measured using rheology at a constant shear rate of 0.1 s−1 and in a temperature range between 0 and 100 °C (Fig. 2). There were significant increases in the dynamic viscosity as hydroxy groups were added to the termini of linear silicones, the magnitudes of which depended both on the number of hydroxy groups and the silicone molecular weight. Even covalently linking as few as two hydroxy groups increased the viscosity at 0 °C by a factor of 50 (SAS9-4), 32 (SAS38-4), 375 (SAS65-4), and 7 (SAS673-4). The increase in viscosity was particularly striking after the attachment of four hydroxy groups on both ends of the silicone polymer, which led to the greatest increase – a factor of 2.6 × 106 – for the shortest silicone chain (SAS9-8); even the longest silicone chain (SAS673-8) saw a 54 fold increase. The attachment of gluconolactone, which added five hydroxy groups on each end, led to solids (SAS9-10, SAS38-10, SAS65-10) or a highly viscous fluid (SAS673-10) with a 2800 fold increase in viscosity compared to the starting material. Similar trends were observed at 25 °C, albeit of slightly lower magnitude.
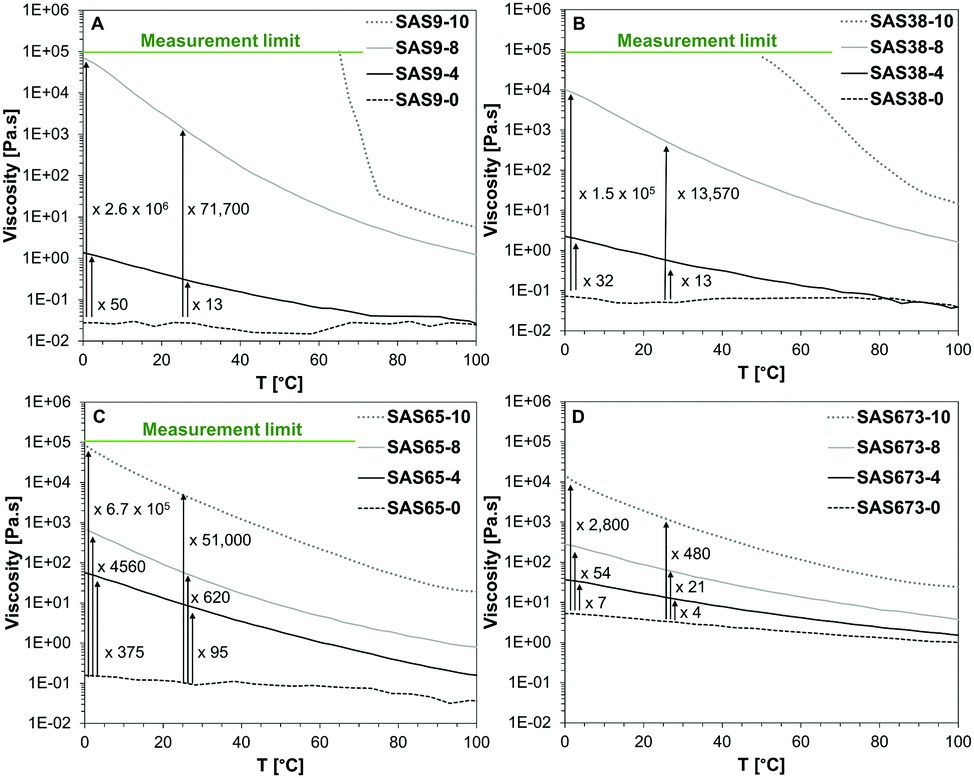 | ||
| Fig. 2 Temperature-dependent dynamic viscosities of aminopropylsilicones (SASn-0) and sugaramidosilicones (SASn-x) with a chain length of n = 9 (A), n = 38 (B), n = 65 (C), and n = 673 (D). The sharp change in the slope of the sample SAS9-10 represents its solidification point. | ||
The influence of an increasing number of hydroxy groups on the dynamic viscosity was not linear and also depended on the silicone chain length of the sugaramidosilicones. The correlation between the number of hydroxy groups and the determined dynamic viscosities is shown for two different temperatures (Fig. 3). The viscosity of the starting aminopropylsilicones (SASn-0) was dependent on the silicone chain length and increased with increasing number of siloxane building units. After the attachment of two hydroxy groups on either side of the chain (SASn-4), however, this trend started to change and after the attachment of four (SASn-8) the trend was inversed – the hydroxy group content became the more important determinant of the dynamic viscosity. The sugaramidosilicone with the shortest silicone chain (SAS9-8) now represented the sample with the highest viscosity, whereas the sugaramidosilicone with the longest silicone chain (SAS673-8) the one with the lowest viscosity (the viscosities of the sugaramidosilicones SAS9-10 and SAS38-10 that bear five hydroxy groups at each terminus were too viscous to be measured at 0 and 25 °C). That is, it is the density of hydroxyl groups [OH]/[Me2SiO] that provides a key element of control with sugaramidosilicones.
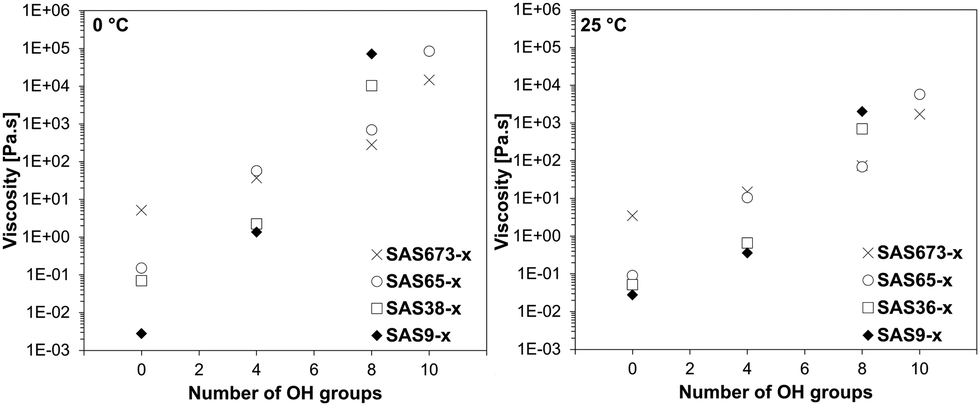 | ||
| Fig. 3 Influence of an increasing number of hydroxy groups on the dynamic viscosity at 0 °C (left) and 25 °C (right) of the respective sugaramidosilicone. | ||
The viscoelastic properties of the fluids and solids were investigated using rheology by the calculation of time-temperature superposition (TTS) curves of constant strain oscillatory frequency measurements at different temperatures (Fig. 4). Note: samples SAS9-x were too viscous to be measured in the same frequency range as the other samples (see Fig. S8, ESI†). Dominant loss moduli demonstrate that the samples have mainly liquid-like properties, whereas dominant storage moduli indicate an elastomer or solid-like state. The intersection of these two, as the angular frequency is increased or decreased, is indicative of a transition of a supramolecular polymer fluid into a solid-state TPN or vice versa.
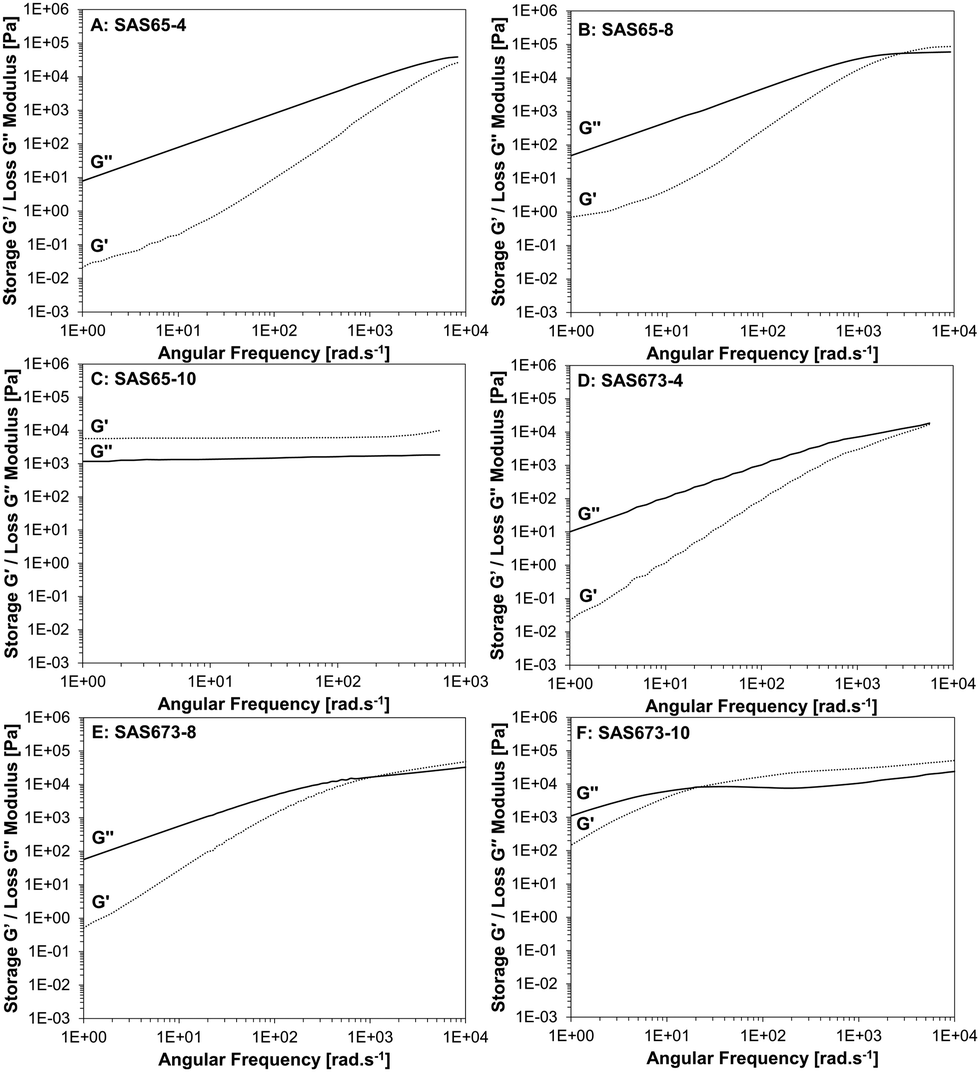 | ||
| Fig. 4 Time-temperature superposition curve of storage G′ and loss G′′ moduli of SAS65-x (A–C), and SAS673-x (D–F). | ||
A distinct gap between the loss and the storage moduli and the absence of an intersection point was observed for the fluid SAS38-4 (for the SAS38-x series see Fig. S9, ESI†). For the samples comprised of longer silicone chains (SAS65-4 and SAS673-4), the storage modulus increased with a higher rate compared to the loss modulus, as the angular frequency was increased leading to converging moduli at high angular frequencies (Fig. 4A and D). An intersection of the storage and loss moduli was observed as the number of hydroxy groups was increased from four to eight (SAS38-8, SAS65-8, and SAS673-8), however, high angular frequencies between 2070 (SAS38-8) and 1130 rad s−1 (SAS673-8) were required. A further increase in the number of hydroxy groups led to the formation of solids (SAS38-10, SAS65-10) for which the storage and loss moduli are frequency independent and the storage modulus dominates over the loss modulus – a characteristic of cross-linked materials. Only sample SAS673-10 remained as a viscous fluid after the modification reaction and the intersection of storage and loss moduli occurred at a significantly lower frequency of 12 rad s−1.
Temperature-dependent dynamic viscosity and constant strain oscillatory frequency rheology measurements indicate a strong dependence of the viscosity and the viscoelastic properties on (i) the silicone chain length, and (ii) the number of covalently attached hydroxy groups. The higher the HO/D unit ratio, the higher the viscosity due to the increased number of intermolecular hydrogen bonds between the sugar-amidosilicone chains. This effect was most distinct for sugaramidosilicones with short silicone chain lengths which transformed from a liquid (SAS9-0, SAS38-0, SAS65-0) to a viscous fluid (SAS9-4, SAS38-4, SAS65-4, SAS9-8, SAS38-8, SAS65-8), and finally to solids (SAS9-10, SAS38-10, SAS65-10).
The behavior of these systems is easily understood by considering the density of hydrogen bonding moieties in the polymer, which will be directly correlated with the chain length of the telechelic polymer, and the effective cross-section of the hydrogen bonding moieties. The latter will be directly related to the number of hydroxy groups at the terminus: 2 vs. 4 vs. 5. Three different regimes present themselves. In the first, a relatively low density of hydrogen bonding moieties leads essentially to chain extension of the polymers that manifests as an increased viscosity (SAS9-4, SAS38-4, SAS65-4, SAS673-4, Fig. 5A). At the other end of the spectrum, the percolation threshold is exceeded either because of many low or fewer high cross-section H-bonding interactions. The product is a dynamic solid (SAS9-10, SAS38-10, SAS65-10, Fig. 5C). Between these two extremes are systems in which the density of H-bonding moieties is sufficiently low that the materials are viscous fluids at rest but form TPNs under compressive or shear force because the H-bonding moieties are forced into effective crosslinks (SAS38-8, SAS65-8, SAS673-8, SAS673-10, Fig. 5B). One can, to a degree, traverse between these systems by heating (Fig. 5C → B or B → A) to overcome some of the H-bonding interactions (Fig. S10 and S11, ESI†).
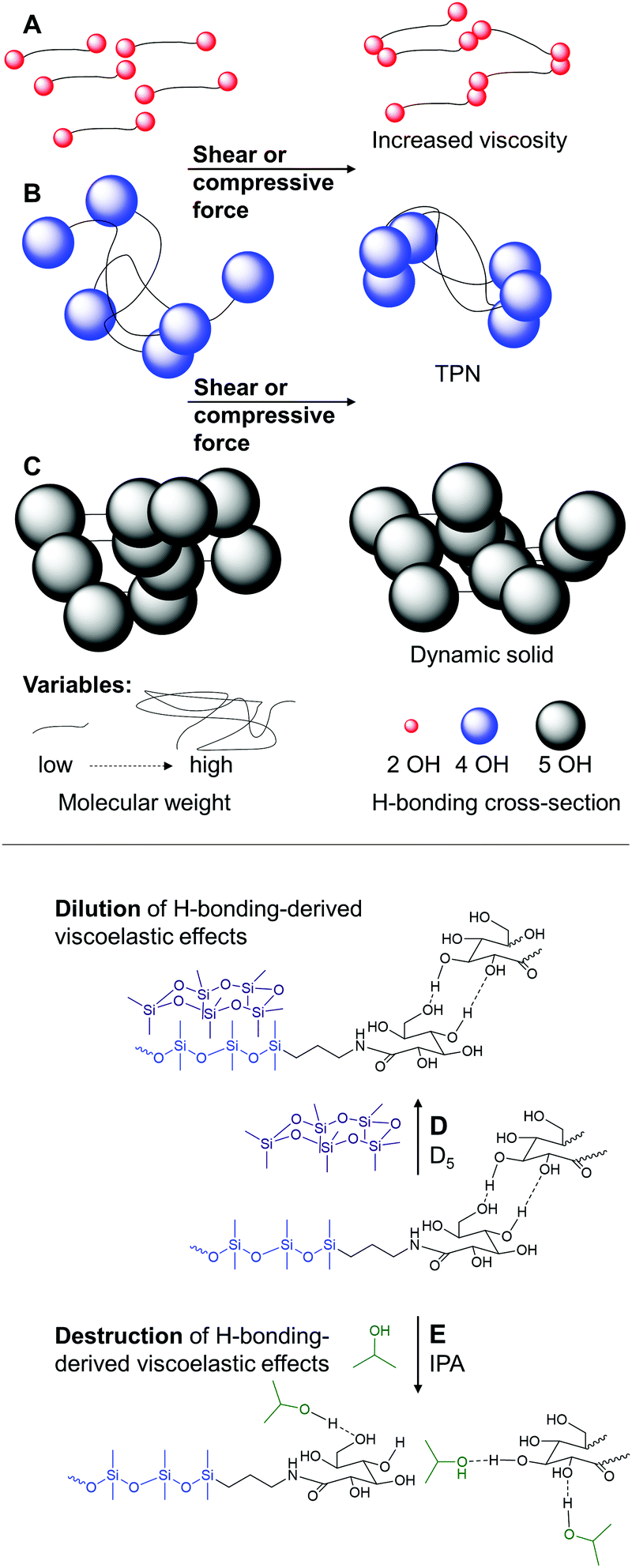 | ||
| Fig. 5 (A) Viscosity increase only due to dynamic H-bonding of sugars. (B) Systems with intermediate overlap between sugars that, under stress, form TPNs. (C) Solid formation due to permanent efficient overlap of H-bonding moieties. Addition of (D) D5 which dilutes the silicone fraction, (E) IPA, which interrupts the hydrogen bonds between sugars. | ||
The density of hydrogen bonding moieties additionally affects the frequency needed for the formation of TPNs. The high molecular sample SAS673-10 forms a TPN at low angular frequencies (12 rad s−1), whereas SAS673-8 with a lower density of hydrogen bonding moieties requires a much higher frequency.
The density of hydrogen bonding moieties can additionally be moderated by the addition of silicone oils. Dilution of solids with the silicone oil D5 decreases the HO/D ratio and converts sugaramidosilicone solids to TPN-forming fluids (Fig. 5C → B), as shown for the sample SAS65-10 by constant strain oscillatory rheology experiments (Fig. S12, ESI†). Dilution of the sugar residue concentration by about 1/10 allows the viscoelastic properties of a 10 wt% solution of SAS65-10 in D5 to converge with the properties of undiluted SAS673-10, which simultaneously proves that the silicone oil does not interfere with hydrogen bonding (Fig. 5D and Fig. S13, ESI†).
Based on the preceding argument, the disruption of hydrogen bonding should preclude the formation of TPNs in these systems. Adding to SAS673-10 even as little as 2 wt% of a good solvent for silicone that is also a good hydrogen bonder – 2-propanol (IPA) – destroyed its ability to form a TPN (Fig. 5E); no intersection between the storage and the loss moduli was observed after the addition of IPA (Fig. S13, ESI†). Similar to its parent sugar-free starting material, SAS673-0, the fluid SAS673-10 diluted with 10 wt% IPA (SAS673-10-IPA-10) cannot absorb energy and thus barely retards motion of a dropped steel ball (Fig. 6, for videos see ESI†). Thus, the hydrogen cross-section can be tuned downwards by the addition of other hydrogen bonding agents that disrupt sugar/sugar crosslinks. An analogous effect was observed, but with lower efficiency, when SAS673-10 was diluted with aprotic, polar THF due to its weak hydrogen bonding character. The addition of up to 5 wt% water has the opposite effect. It strengthens hydrogen bonding and shifts the frequency needed for the formation of TPNs to lower values (Fig. S13, ESI†).
 | ||
| Fig. 6 Location (0.5 s after impact) of a 12 mm steel ball (m = 6.95 g) dropped from 10 cm height onto: SAS673-10 (a), SAS673-10-D5-10 (b), SAS673-THF-10 (c), SAS673-10-IPA-10 (d), and SAS673-0 (e). The sample height was 6 mm. | ||
In conclusion, supramolecular silicones can be formed by the covalent attachment of hydroxy groups to linear silicone chains. Intermolecular hydrogen bonds between the hydroxy groups increase the interaction of adjacent sugaramidosilicones compared to their unmodified starting materials thus increasing the dynamic viscosities. Both silicone molecular weight and the number of available HO groups define visco-elastic properties, including the ability to form TPNs under shear. These interactions can be tuned or overcome by addition of hydrogen bonding solvents, leading to dramatic reduction in viscosity and energy absorption abilities which is highly interesting in the field of personal protective applications such as liquid armor.
The authors gratefully acknowledge the Austrian Science Fund (FWF) (Proj. Nr J 4217-N28, A. F.) and the NSERC Canada (MAB) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS.
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922–13923 CrossRef CAS.
- J. R. Kumpfer, J. Jin and S. J. Rowan, J. Mater. Chem., 2010, 20, 145–151 RSC.
- A. Noro, S. Matsushima, X. He, M. Hayashi and Y. Matsushita, Macromolecules, 2013, 46, 8304–8310 CrossRef CAS.
- T. Rossow, A. Habicht and S. Seiffert, Macromolecules, 2014, 47, 6473–6482 CrossRef CAS.
- S. Suzuki, T. Uneyama, T. Inoue and H. Watanabe, Macromolecules, 2012, 45, 888–898 CrossRef CAS.
- Y. Séréro, V. Jacobsen, J.-F. Berret and R. May, Macromolecules, 2000, 33, 1841–1847 CrossRef.
- H. Bagger-Jörgensen, L. Coppola, K. Thuresson, U. Olsson and K. Mortensen, Langmuir, 1997, 13, 4204–4218 CrossRef.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS.
- J. R. Perkins, I. Diboun, B. H. Dessailly, J. G. Lees and C. Orengo, Structure, 2010, 18, 1233–1243 CrossRef CAS.
- F. Vidal, J. Gomezcoello, R. A. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2019, 141, 15963–15971 CrossRef CAS.
- A. Jangizehi, S. R. Ghaffarian, W. Schmolke and S. Seiffert, Macromolecules, 2018, 51, 2859–2871 CrossRef CAS.
- M. Golkaram and K. Loos, Macromolecules, 2019, 52, 9427–9444 CrossRef CAS.
- C. L. Lewis, K. Stewart and M. Anthamatten, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
- J. K. Hirschberg, F. H. Beijer, H. A. van Aert, P. C. Magusin, R. P. Sijbesma and E. Meijer, Macromolecules, 1999, 32, 2696–2705 CrossRef.
- B. J. Folmer, R. Sijbesma, R. Versteegen, J. Van der Rijt and E. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- L. Sangroniz, A. Sangroniz, A. Alegria, M. Fernandez, L. Irusta, A. J. Müller, A. Etxeberria and A. Santamaría, Polymer, 2018, 159, 12–22 CrossRef CAS.
- L. Yang, Y. Lin, L. Wang and A. Zhang, Polym. Chem., 2014, 5, 153–160 RSC.
- A. S. Fawcett and M. A. Brook, Macromolecules, 2014, 47, 1656–1663 CrossRef CAS.
- L. Dodge, Y. Chen and M. A. Brook, Chem. – Eur. J., 2014, 20, 9349–9356 CrossRef CAS.
- K. Faiczak, M. A. Brook and A. Feinle, Macromol. Rapid Commun., 2020, 2000161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Contains full experimental, characterization, rheology, DSC, and ball dropping experiment data. See DOI: 10.1039/d0cc05478j |
| This journal is © The Royal Society of Chemistry 2020 |