
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetic selection of Pd4L2 metallocyclic and Pd6L3 trigonal prismatic assemblies†
Helen M.
O’Connor
 ,
Marco
Coletta
,
Alvaro
Etcheverry-Berríos
,
Gary S.
Nichol
,
Euan K.
Brechin
* and
Paul J.
Lusby
*
,
Marco
Coletta
,
Alvaro
Etcheverry-Berríos
,
Gary S.
Nichol
,
Euan K.
Brechin
* and
Paul J.
Lusby
*
EaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: E.Brechin@ed.ac.uk; Paul.Lusby@ed.ac.uk
First published on 25th August 2020
Abstract
The self-assembly of Pd4L2 metallocylcic and Pd6L3 trigonal prismatic assemblies are described. The selection of one species over the other has been achieved by careful choice of ancilliary ligands, which switch the dynamics of the Pd-pyridine bonds such that a highly unusual and distorted smaller assembly can be kinetically trapped en route to the more energetically favourable larger species. Both assemblies provide promise as easy to access multicavity reaction vessels.
The ability to exploit synthetic capsules to mimic complex biological systems has led to the development of a breadth of beautiful and complex systems including M4L6 tetrahedra, M8L12 cubes, and M12L24 nanospheres.1–3 Many times these species are obtained as the thermodynamic product by carefully designing a multitopic ligand, which when combined with a metal ion of specific geometry, yields the predicted structure in very high yield. Less frequent are those assemblies that represent a kinetic trap on the potential energy surface, which usually occurs with a less dynamic transition metal ion e.g. Pt2+ instead of Pd2+ (or Ru2+ instead of Fe2+).4,5 Structures that incorporate a cis-protected square-planar transition metal ion component, which followed on from Fujita's breakthrough report of a molecular square in 1990,6 provide a different way to tune the system dynamics – by changing the ancillary bidentate ligand. These can provide either a strong or weak trans-effect, which can tune the metal-multitopic ligand interactions from labile to more inert. While there have been numerous separate reports that involve assemblies with either strong trans-effect ancillary ligands, such as the diphosphines commonly utilised by Stang and others,7–10 or weaker trans-effect ligands, usually nitrogen donors such as ethylene diamine and their derivatives,6,11,12 studies that involve and simultaneously compare the two are relatively scarce.13–15 In this study we provide a rare direct comparison, and show how careful selection can be used to control the formation of different architectures.
The initial motivation behind this research was not to reveal the intricacies of self-assembly through control of kinetic factors, but rather to create new reaction vessels for bio-inspired catalysis. We have recently shown that Pd2L4 “molecular lantern” assemblies, C-1 and C-2 (Fig. 1a), can be remarkably effective catalysts, combining high activity with facile turnover.16–18 Moreover, we have also been interested in creating systems that provide a more open cavity, such as Pd2L2 hemi-cage, HC (Fig. 1b),19 which would permit better access to reactants and thus widen the substrate scope. We were also interested in creating structures which possess the features of the hemi-cage system i.e. the more accessible pseudo cavity that resides between the two Pd2+ ions, but are better pre-organised (Fig. 1c). These systems also possess a secondary more hydrophobic central cavity that could bind an additional less polar guest. Multicavity systems provide further interest because they represent a route into allosterically regulated catalysis,20 however, the synthesis of these systems are often hindered by complex ligand-design,21–25 or rely on the serendipitous formation of interlocked sub-structures.26,27 The approach we envisaged here can be considered much simpler as it involves the use of the easy to access tetratopic ligand, 1,2,4,5-tetra(pyridin-3-ylethynyl)benzene (LT), which is one step from commercially available starting materials (see, ESI†).
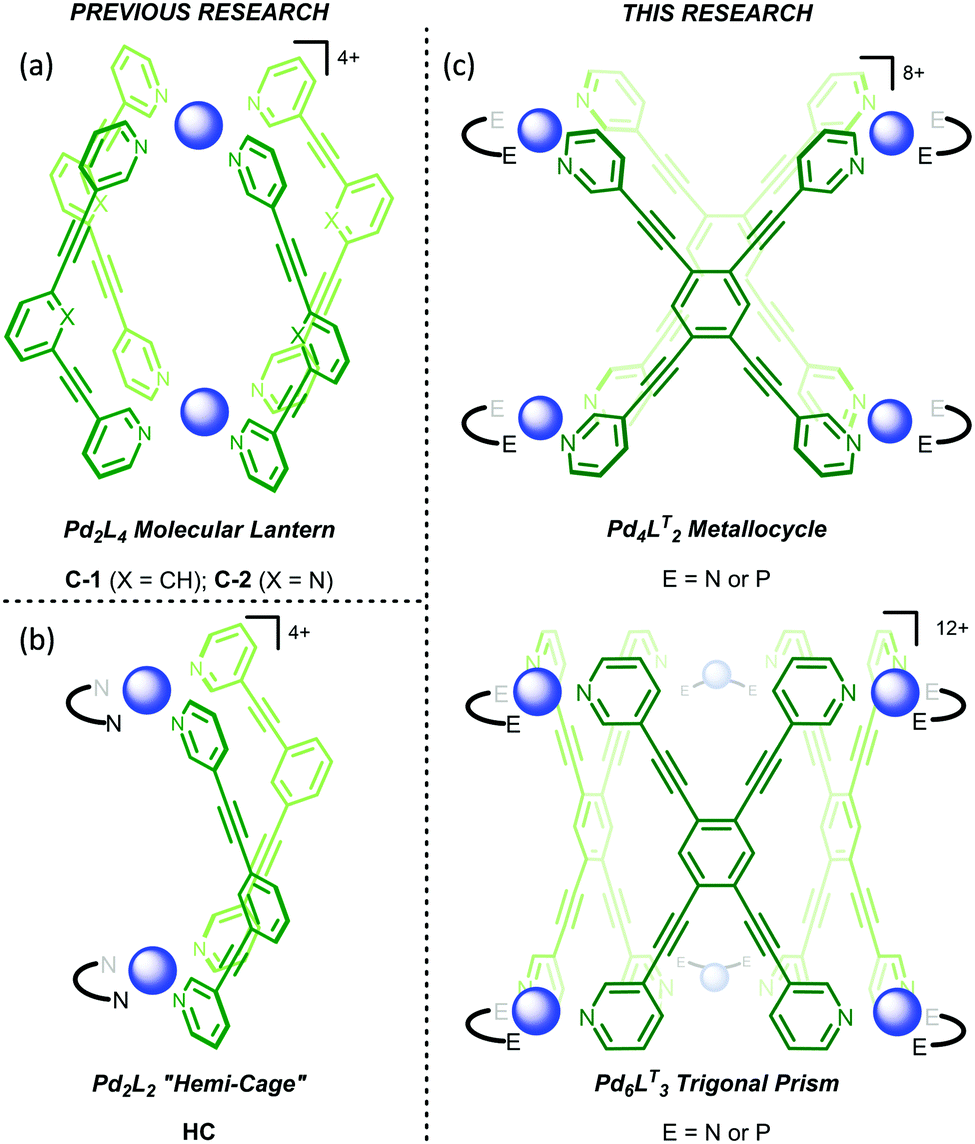 | ||
| Fig. 1 Structural representation of the (a) molecular lanterns C-1 (X = CH) and C-2 (X = N), (b) “hemi-cage” HC, and (c) Pd4LT2 metallocycle (top) and Pd6LT3 trigonal prism (bottom; E = N or P). The Pd2+ ions are represented by the blue spheres. | ||
Combining a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of [(TMEDA)Pd(OTf)2] (TMEDA = tetramethylethylenediamine) and LT in nitromethane leads to the immediate formation (<5 min) of one set of dominant 1H NMR signals (Fig. 2, 0 hours, orange resonances) that possess the same symmetry as the free ligand. This is followed by the gradual appearance of a second set of resonances with the same symmetry (Fig. 2, 12 h, green resonances). Over a period of 2 days, the first set of signals are almost completely replaced by the second (Fig. 2, 12–48 h). Considering that previous work has shown that rigid tetratopic ligands can generate M6L3 trigonal prismatic and M8L4 tetragonal prismatic structures when reacted with cis-protected square planar complexes,28–32 and that even higher order species can form,33 we initially attributed these two sets of peaks to the Pd′6LT3 and Pd′8LT4 structures (Pd′ = (TMEDA)Pd). We were surprised, however, to discover that single crystals grown from diffusion of diethyl ether into the reaction mixture revealed the highly strained Pd′4LT2 structure,34,351a·OTf (Fig. 3 and Fig. S4.5.1, ESI†).
:1 ratio of [(TMEDA)Pd(OTf)2] (TMEDA = tetramethylethylenediamine) and LT in nitromethane leads to the immediate formation (<5 min) of one set of dominant 1H NMR signals (Fig. 2, 0 hours, orange resonances) that possess the same symmetry as the free ligand. This is followed by the gradual appearance of a second set of resonances with the same symmetry (Fig. 2, 12 h, green resonances). Over a period of 2 days, the first set of signals are almost completely replaced by the second (Fig. 2, 12–48 h). Considering that previous work has shown that rigid tetratopic ligands can generate M6L3 trigonal prismatic and M8L4 tetragonal prismatic structures when reacted with cis-protected square planar complexes,28–32 and that even higher order species can form,33 we initially attributed these two sets of peaks to the Pd′6LT3 and Pd′8LT4 structures (Pd′ = (TMEDA)Pd). We were surprised, however, to discover that single crystals grown from diffusion of diethyl ether into the reaction mixture revealed the highly strained Pd′4LT2 structure,34,351a·OTf (Fig. 3 and Fig. S4.5.1, ESI†).
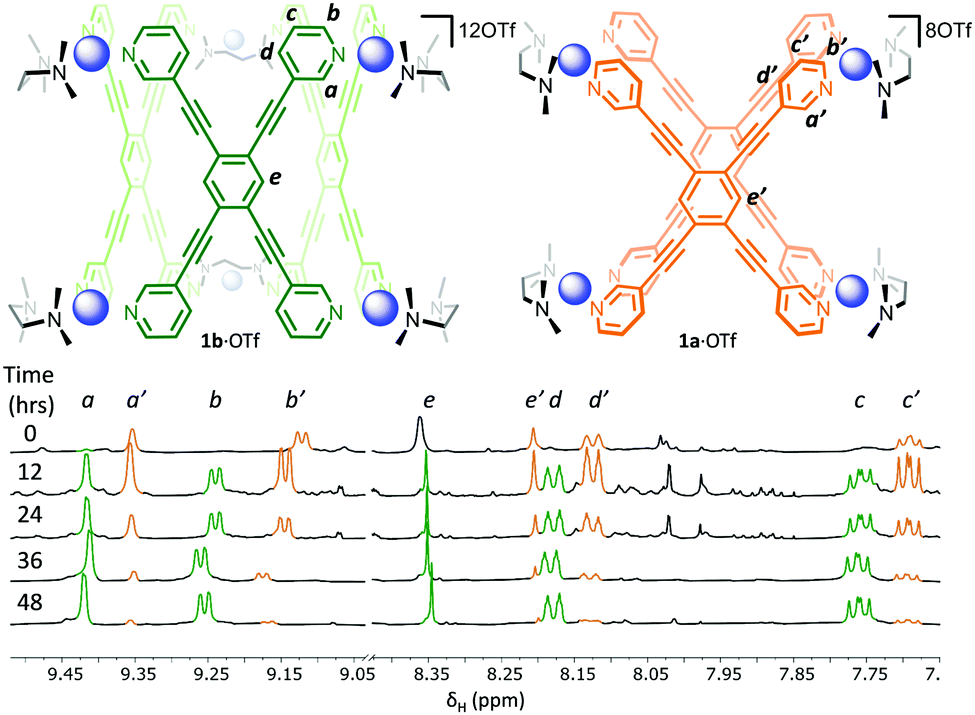 | ||
| Fig. 2 Time dependent 1H NMR spectra (500 MHz, CD3NO2, 300 K) showing the initial formation of 1a·OTf (orange signals) followed by the conversion into 1b·OTf (green signals) over a period of 48 hours. | ||
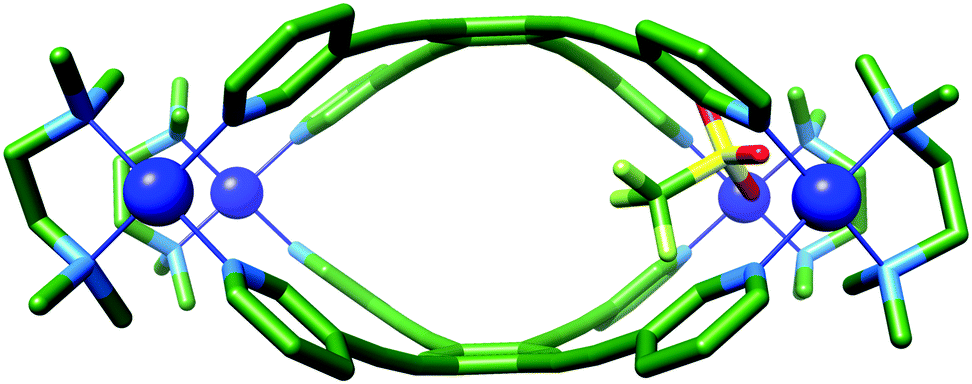 | ||
| Fig. 3 X-ray crystal structure of 1a·OTf. Protons and additional counterions have been removed for clarity. Colour code: C: green, N: light blue, O: red, S: yellow, P: orange, Pd: blue, F: light green. | ||
The molecular structure of 1a·OTf can be described as two portals linked by a very small central cavity that is formed from just two ligands which display significant curvature away from planarity. Each portal is defined by two palladium ions linked by the 1,3-bispyridyl-motif. The Pd–Pd distances in these two portals are 9.5 Å, which is considerably shorter than the ca. 12 Å that is observed for C-1/C-2, even though the supposedly rigid (pyridin-3-ylethynyl)benzene linker between the two metal ions is the same. Unlike C-1/C-2, where there is little distortion of the ligand or metal, 1a8+ shows significant deformation of both components. This is particularly evident in LT, where the angle between the planes of the coordinating pyridyl rings in the free ligand and 1a8+ distort from 125° to 104°, respectively (Fig. S4.4, ESI†). This bending and contraction of the portal Pd–Pd distance indicates that closure of the structure to give 1a8+ requires significant distortion. The Pd2+ ions of 1a8+ are also slightly deviated away from square planar geometry with NL–Pd–NL and NTMEDA–Pd–NTMEDA, and NL–Pd–NTMEDA angles approximately 87.0° and 93.2°, respectively. One of the portals is occupied by a charge balancing triflate ion, which shows close contacts to eight inward facing C–H bonds (Ar–H⋯O distances: 2.4–3.2 Å); four each from the methyl groups of the TMEDA ligand and the o-pyridyl hydrogen atoms of LT. These C–H bonds are polarised by their proximity to the charged Pd2+ ion, and thus become reasonable H-bond donors. The remaining seven charge balancing triflate ions are located outside of the cavities and contact the extended structure via a number of interactions, the shortest of which is 2.3 Å (py–H⋯O; Fig. S4.6.1, ESI†).
In order to confirm that the species formed in solution is Pd′4LT2, and to ascertain the identity of the second species, we further investigated using both ESI-MS and 1H DOSY NMR spectroscopy. As expected, ESI-MS (Fig. S3.1.1 and S3.1.2, ESI†) shows the charge states for the Pd′4LT2 species (3+ and 4+), but also the presence of a Pd′6LT3 species, 1b·OTf (4+ and 5+). There is no evidence of any higher order species. Taken in conjunction with the change in the 1H NMR spectra as a function of time (Fig. 2), this indicates that 1a8+ is initially formed before slowly converting into 1b12+. The 1H DOSY NMR spectrum also shows that the two species are similar in size (Fig. S2.2.2, ESI†) and that their hydrodynamic radii, calculated using the Stokes–Einstein equation, is ca. 12 Å. Molecular models of Pd′6LT3 and Pd′8LT4 were generated using Spartan 10′ (Fig. S5.1, ESI†), and their maximum diameter was examined, along with 1a8+, using the program pywindow.36 The maximum diameter is defined as the distance between the edges of the van der Waals spheres of the two atoms at the greatest distance from each other in the molecule. Diameters of 25, 27, and 31 Å were extracted for 1a8+, 1b12+, and the tetragonal prism, respectively. Negating solvent and/or anion effects, these diameters are closest to the two smaller species, indicating that the solution assemblies are indeed 1a·OTf and 1b·OTf. These results further support that 1a·OTf is a highly strained kinetic intermediate, and 1b·OTf is the more thermodynamically stable final assembly.
The effect that the cis-capping ligand has on the self-assembly process is quite pronounced, as evidenced by changing TMEDA to diphenylphosphinopropane (dppp). The combination of LT with [(dppp)Pd(OTf)2] in a 1:2 ratio in acetonitrile gave a 1H NMR spectrum that showed a single high symmetry species with slightly broadened signals, which significantly showed no change over 3 days (Fig. S2.2.5, ESI†). Complexation of LT is evidenced by the characteristic shift of the pyridyl signals and by the splitting of the propyl resonances of the dppp ligand (Fig. S2.2.4a, ESI†). ESI-MS again showed Pd′′4LT2 (Pd′′ = (dppp)Pd) and Pd′′6LT3 species, with no evidence of larger assemblies (Fig. S3.2.1 and S3.2.2, ESI†). The observation of different speciation by NMR spectroscopy and MS can likely be explained by fragmentation of Pd′′6LT3 under ionising conditions. The direct formation of Pd′′6LT3 in solution would also be consistent with the trans-labilising properties of the dppp ligand, which would render any highly distorted Pd′′4LT2 intermediate kinetically unstable, thus promoting rapid navigation towards the energy minima on the potential energy surface.
Single X-ray quality crystals could not be grown directly from the self-assembly reaction of LT with [(dppp)Pd(OTf)2]. Instead, the product was first isolated as a powder by precipitation of a concentrated sample with diethyl ether, and subsequently crystallised using diethyl ether vapour diffusion into an acetonitrile solution yielding a small amount of crystals suitable for single crystal X-ray diffraction (Fig. S4.10, ESI†). X-ray crystallographic analysis from data collected with synchrotron radiation was used to confirm the Pd′′6LT3 trigonal prism structure, 2·OTf,37 with three triflate-containing portals (Fig. 4 and Fig. S4.5.3, ESI†). There are several short host–guest contacts, similar to 1a·OTf, between the triflate O atoms and the polarised C–H bonds of both the capping and bridging ligand. In addition, the fluorine atoms of the triflate CF3 group are very close to the central CH of LT with Ar–H⋯F distances approximately 2.2 Å. The remaining charge balancing triflate ions provide numerous electrostatic interactions between the neighbouring trigonal prisms, the shortest of which is between the O atom of the triflate ion and the outer o-pyridyl H-bond donor pockets (2.3 Å; Fig. S4.6.2, ESI†)
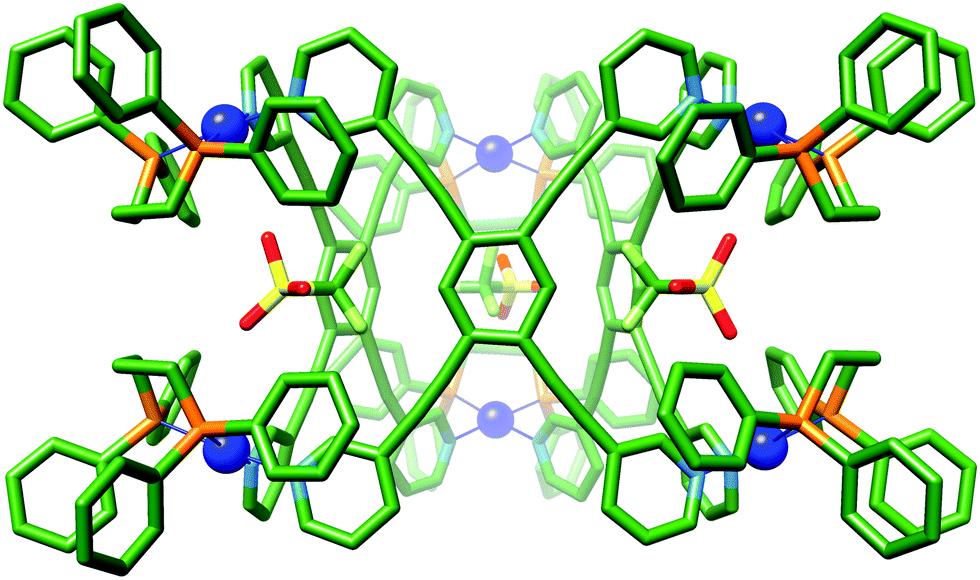 | ||
| Fig. 4 X-ray crystal structure of 2·OTf. Protons and additional counterions have been removed for clarity. Colour code: C: green, N: light blue, O: red, S: yellow, P: orange, Pd: blue, F: light green. | ||
In order to examine the encapsulation of organic guests in solution, with a view to conducting catalytic studies, we attempted to exchange the triflate ions for tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF−). We have previously shown that these non-interacting anions leave the polar surfaces of cages exposed, promoting the binding of polar organic guests. Unfortunately, efforts to react 1·OTf with NaBArF were unsuccessful, and only partial exchange of the triflate ion could be achieved (Fig. S2.2.6, ESI†). In contrast, a similar experiment with 2·OTf and NaBArF gave a product, which when analysed by 1H NMR spectroscopy showed a single species with sharp resonances that was consistent with retention of the cage structure (Fig. 5). The 19F NMR spectrum also showed almost full consumption of the OTf− signal, with only trace amounts present. The 1H DOSY NMR spectrum gave an increased hydrodynamic radius of 19 Å, in line with calculated values (Fig. S2.2.7, ESI†), and consistent with the successful conversion of 2·OTf to 2·BArF.
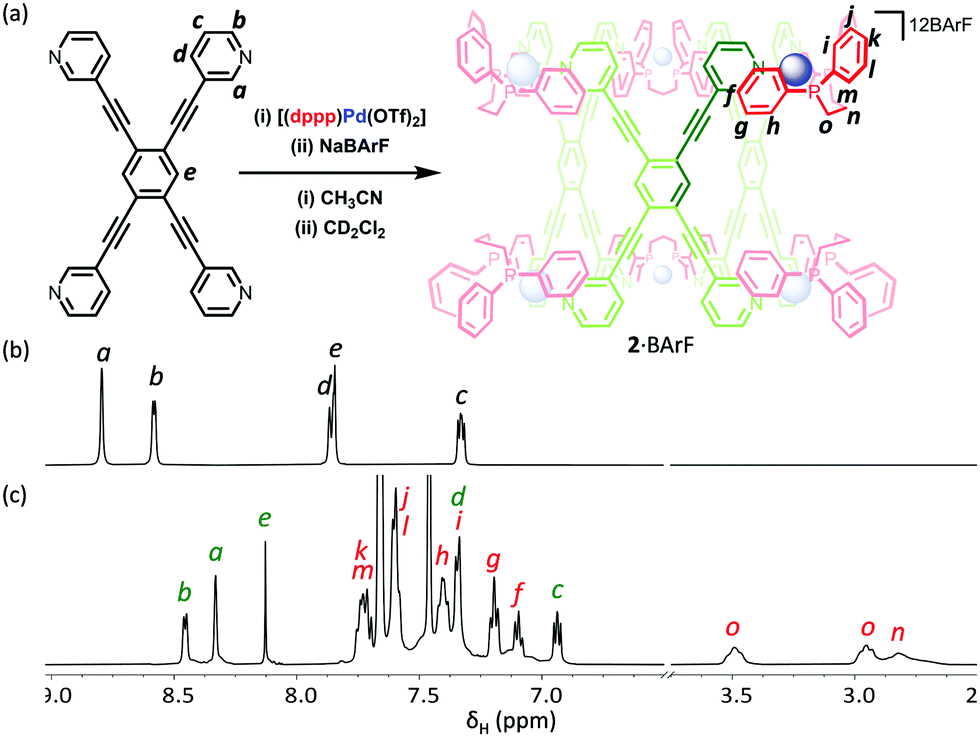 | ||
| Fig. 5 (a) Schematic representation of the preparation of 2·BArF via (i) the self-assembly of LT and [(dppp)Pd(OTf)2] and (ii) anion exchange with NaBArF; 1H NMR spectra (500 MHz, CD2Cl2, 300 K) of (b) LT and (c) 2·BArF. | ||
Initial binding studies were performed using a series of quinones (benzo-, naphtho- and anthraquinone) in CD2Cl2. We have previously shown that these bind to both C-1/C-2 and HC systems through hydrogen bonding of the carbonyl groups to the polarised CH bonds that are in close proximity to the Pd2+ ion. While the crystal structure of 2·OTf revealed that the portal Pd–Pd distances were shorter than either of these systems, we were hopeful that 212+ would be able to flex sufficiently to accommodate these guests. However, we were disappointed by the negligible shifts in the 1H NMR spectra associated by both the inward facing o-pyridyl (Ha) and central (He) protons (Fig. S6.2, ESI†), which indicates that these quinones are not encapsulated. We have also tried guests that have their carbonyl groups more closely spaced than that of the quinones (e.g. cyclopentenedione) and also single carbonyl species (e.g. chromone) but only minimal evidence of ingress could be observed (Fig. S6.3 and S6.4, ESI†). We attribute the lack of interior binding to two possible factors, which are not mutually exclusive. Firstly, the connection of the three portals rigidifies the overall structure so that it cannot “breathe” in order to accommodate these guests. Secondly, there is poor access to the portal either directly past the bulky dppp ligands (Fig. S4.5.4, ESI†) or via the central cavity. Interestingly, chromone shifts some of the resonances on the outside of the cage (Hb), with the signals of chromone also broadening quite noticeably (Fig. S6.4, ESI†), which indicates that this species may interact with the outer pocket of H-bond donors. We have previously seen a similar binding mode with C-1/C-2 and Ph3PO.38 This provides further evidence that encapsulation is prohibited because of thermodynamic (i.e. requiring significant cage distortion) and/or kinetic (i.e. poor access) factors.16,19
In conclusion, we have shown how different multi-component assemblies can be made using cis-capping ligands to tune the reversibility of M–L interactions, facilitating access to both kinetic and thermodynamic products. We continue the search for suitable guests that will allow the overall goal of creating simple, multi-cavity systems, in which catalysis can be controlled via allosteric binding.
We thank the EPSRC (H. O. C., EP/P025986/1EP; M. C. EP/N01331X/1) and ANID (Chile) for a Postdoctoral Fellowship (A. E. B. grant number 74190043). We thank Diamond Light Source for access to synchrotron facilities, award CY22240. GSN thanks William Clegg, Paul G. Waddell and Michael R. Probert for their support with synchrotron data collection. We acknowledge the NMR, MS, and X-ray departments at EaStCHEM and thank Rebecca Spicer and Dr Claire Hobday for their advice and assistance.
Conflicts of interest
There are no conflicts to declare.References
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- M. Fujita, F. Ibukuro, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175–4176 CrossRef CAS.
- S. Roche, C. Haslam, S. L. Heath and J. A. Thomas, Chem. Commun., 1998, 1681–1682 RSC.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS.
- P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981–4982 CrossRef CAS.
- M. Hardy, N. Struch, J. J. Holstein, G. Schnakenburg, N. Wagner, M. Engeser, J. Beck, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2020, 59, 3195–3200 CrossRef CAS.
- N. J. Cookson, J. M. Fowler, D. P. Martin, J. Fisher, J. J. Henkelis, T. K. Ronson, F. L. Thorp-Greenwood, C. E. Willans and M. J. Hardie, Supramol. Chem., 2018, 30, 255–266 CrossRef CAS.
- R. Pinalli, V. Cristini, V. Sottili, S. Geremia, M. Campagnolo, A. Caneschi and E. Dalcanale, J. Am. Chem. Soc., 2004, 126, 6516–6517 CrossRef CAS.
- E. Holló-Sitkei, G. Tárkányi, L. Párkányi, T. Megyes and G. Besenyei, Eur. J. Inorg. Chem., 2008, 1573–1583 CrossRef.
- N. B. Debata, D. Tripathy and D. K. Chand, Coord. Chem. Rev., 2012, 256, 1831–1945 CrossRef CAS.
- S. Ghosh and P. S. Mukherjee, Inorg. Chem., 2009, 48, 2605–2613 CrossRef CAS.
- Z. Qin, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2003, 42, 1956–1965 CrossRef CAS.
- L. Pirondini, F. Bertolini, B. Cantadori, F. Ugozzoli, C. Massera and E. Dalcanale, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4911–4915 CrossRef CAS.
- V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef.
- T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, J. Am. Chem. Soc., 2020, 142, 1300–1310 CrossRef.
- R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS.
- V. Martí-Centelles, F. Duarte and P. J. Lusby, Isr. J. Chem., 2019, 59, 257–266 CrossRef.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS.
- K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914–15921 CrossRef CAS.
- P. N. W. Baxter, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Chem. – Eur. J., 1999, 5, 113–120 CrossRef CAS.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS.
- S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem. – Eur. J., 2014, 20, 13122–13126 CrossRef CAS.
- M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem. – Eur. J., 2015, 21, 3948–3955 CrossRef CAS.
- S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef.
- Y.-H. Li, J.-J. Jiang, Y.-Z. Fan, Z.-W. Wei, C.-X. Chen, H.-J. Yu, S.-P. Zheng, D. Fenske, C.-Y. Su and M. Barboiu, Chem. Commun., 2016, 52, 8745–8748 RSC.
- Y. Yamanoi, Y. Sakamoto, T. Kusukawa, M. Fujita, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2001, 123, 980–981 CrossRef CAS.
- D. C. Caskey, T. Yamamoto, C. Addicott, R. K. Shoemaker, J. Vacek, A. M. Hawkridge, D. C. Muddiman, G. S. Kottas, J. Michl and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 7620–7628 CrossRef CAS.
- S. C. Johannessen, R. G. Brisbois, J. P. Fischer, P. A. Grieco, A. E. Counterman and D. E. Clemmer, J. Am. Chem. Soc., 2001, 123, 3818–3819 CrossRef CAS.
- P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS.
- A. K. Bar, S. Mohapatra, E. Zangrando and P. S. Mukherjee, Chem. – Eur. J., 2012, 18, 9571–9579 CrossRef CAS.
- G. Cecot, M. Marmier, S. Geremia, R. De Zorzi, A. V. Vologzhanina, P. Pattison, E. Solari, F. Fadaei Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2017, 139, 8371–8381 CrossRef CAS.
- Q. Gan, T. K. Ronson, D. A. Vosburg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 1770–1773 CrossRef CAS.
- S. Bivaud, S. Goeb, V. Croué, P. I. Dron, M. Allain and M. Sallé, J. Am. Chem. Soc., 2013, 135, 10018–10021 CrossRef CAS.
- M. Miklitz and K. E. Jelfs, J. Chem. Inf. Model., 2018, 58, 2387–2391 CrossRef CAS.
- N. T. Johnson, P. G. Waddell, W. Clegg and M. R. Probert, Crystals, 2017, 7, 360–369 CrossRef.
- V. Martí-Centelles, R. L. Spicer and P. J. Lusby, Chem. Sci., 2020, 11, 3236–3240 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2007850–2007852. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05311b |
| This journal is © The Royal Society of Chemistry 2020 |