
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A curious case of dynamic disorder in pyrrolidine rings elucidated by NMR crystallography†
Patrick M. J.
Szell
 *a,
Steven P.
Brown
*a,
Leslie P.
Hughes
*b,
Helen
Blade
*b and
Sten O.
Nilsson Lill
*c
*a,
Steven P.
Brown
*a,
Leslie P.
Hughes
*b,
Helen
Blade
*b and
Sten O.
Nilsson Lill
*c
aDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Patrick.Szell@warwick.ac.uk; S.P.Brown@warwick.ac.uk
bOral Product Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield, UK. E-mail: Les.Hughes2@astrazeneca.com; Helen.Blade@astrazeneca.com
cEarly Product Development and Manufacturing, Pharmaceutical Sciences, R&D, AstraZeneca, Gothenburg, Sweden. E-mail: Sten.Nilsson-Lill@astrazeneca.com
First published on 13th October 2020
Abstract
A pharmaceutical exhibits differing dynamics in crystallographically distinct pyrrolidine rings despite being nearly related by symmetry, with one performing ring inversions while the other is constrained to torsional librations. Using 13C solid-state magic-angle spinning (MAS) NMR and DFT calculations, we show that this contrast originates from C–H⋯H–C close contacts and less efficient C–H⋯π intermolecular interactions observed in the transition state of the constrained pyrrolidine ring, highlighting the influence of the crystallographic environment on the molecular motion.
Pharmaceutical products are most often manufactured in their solid forms, benefiting the patient with a convenient route of administration.1 During the development stage, solid forms are thoroughly characterized in order to identify potential risks associated with stability, polymorphic conversion,2,3 and the ability to form hydrates or solvates.4 Characterization may include X-ray crystallography and using the derived structural model to assess the risks of making a particular solid form into a medicine. The occurrence of crystallographic disorder arising from mobility (i.e. dynamic structural disorder) or the accessibility of multiple conformations/orientations (i.e. static structural disorder) poses several challenges in the risk assessment, due in part to the uncertainty on the atomic positions.
Solid-state NMR spectroscopy is a powerful tool for investigating crystallographic disorder, with the potential to exploit several pharmaceutically-relevant nuclei (1H, 13C, 15N) and the ability to probe specific sites in the structure.5,6 NMR crystallography is capable of distinguishing static from dynamic structural disorder, has been used for investigating dynamics in pharmaceuticals,7–10 and can be used to improve structural models.11–17 Conversely, the presence of dynamics may not be immediately apparent from X-ray data, especially for data acquired at low temperatures due to a “freezing” of the motion.
Here, we combine solid-state NMR and DFT calculations in an NMR crystallography approach to investigate a development compound, 1a, which features a curious case of structural disorder. Despite there being two molecules in the asymmetric unit (Z′ = 2) each related by pseudosymmetry, surprisingly only one of the two pyrrolidine groups in the structural model appears to be disordered. The solid-state NMR experiments allow the motion and thermodynamic parameters to be characterized in 1avia13C spin–lattice relaxation time measurements, while the computations allow the origins of these contrasting dynamics to be understood.
The compound investigated herein, 1a, consists of the salt (the counterion is referred to as “a”) of a pharmaceutical compound (1) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric equivalence. The structural model, determined by X-ray crystallography at 150 K, suggests the presence of a pair of 1a related by C2 pseudosymmetry, with the disorder in one of the pyrrolidine groups of 1 breaking this symmetry. As shown in Fig. 1a, where the red dashed lines represent the rest of the undisclosed structure, a pyrrolidine group appears to be relatively “ordered” (henceforth referred to as Cord), while the other group appears to be disordered (henceforth referred to as Cdis) over two positions with occupancies of 0.5 each. However, while Cord appears to be ordered, the situation is ambiguous as its anisotropic displacement ellipsoids have some distortions (see Fig. S3 of the ESI†), suggesting the presence of vibrations. The crystallographic environment surrounding the two pyrrolidine groups differ in that Cdis interacts more closely with the counterion a while Cord interacts primarily with other molecules of 1. All contacts (within 3 Å) involving the pyrrolidine groups are shown in Fig. S4 of the ESI.† In order to confirm the contrast in the dynamics of the pyrrolidine groups, variable temperature 1H–13C cross polarisation (CP) magic-angle spinning (MAS) solid-state NMR experiments and 13C spin–lattice relaxation time measurements, T1(13C), have been performed.
:1 stoichiometric equivalence. The structural model, determined by X-ray crystallography at 150 K, suggests the presence of a pair of 1a related by C2 pseudosymmetry, with the disorder in one of the pyrrolidine groups of 1 breaking this symmetry. As shown in Fig. 1a, where the red dashed lines represent the rest of the undisclosed structure, a pyrrolidine group appears to be relatively “ordered” (henceforth referred to as Cord), while the other group appears to be disordered (henceforth referred to as Cdis) over two positions with occupancies of 0.5 each. However, while Cord appears to be ordered, the situation is ambiguous as its anisotropic displacement ellipsoids have some distortions (see Fig. S3 of the ESI†), suggesting the presence of vibrations. The crystallographic environment surrounding the two pyrrolidine groups differ in that Cdis interacts more closely with the counterion a while Cord interacts primarily with other molecules of 1. All contacts (within 3 Å) involving the pyrrolidine groups are shown in Fig. S4 of the ESI.† In order to confirm the contrast in the dynamics of the pyrrolidine groups, variable temperature 1H–13C cross polarisation (CP) magic-angle spinning (MAS) solid-state NMR experiments and 13C spin–lattice relaxation time measurements, T1(13C), have been performed.
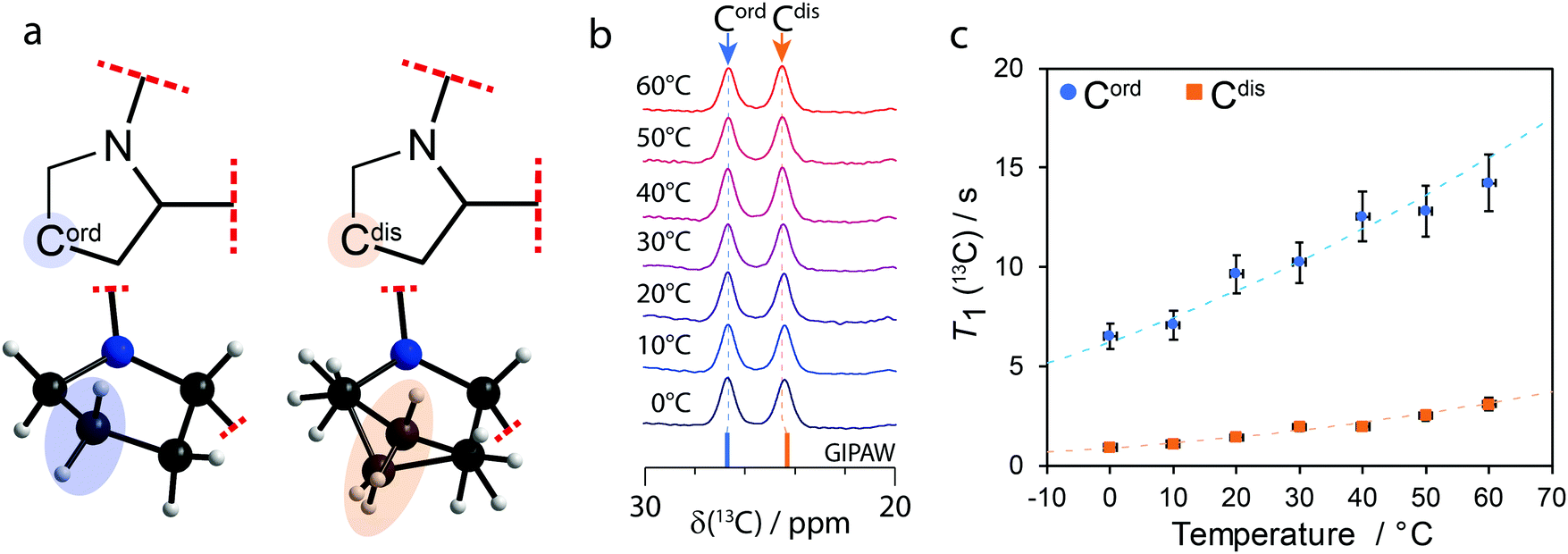 | ||
| Fig. 1 (a) Diagram of the partial molecular structure and depiction of the structural model of 1a showing the ordered (Cord) and disordered (Cdis) carbon atoms on their respective pyrrolidine groups. (b) Variable temperature 1H–13C CPMAS (νL(1H) = 400 MHz; νMAS = 10 kHz) solid-state NMR spectra of 1a showing the 20 to 30 ppm region and (c) T1(13C) of Cord and Cdis as a function of temperature. The dashed lines in (c) show the fits using eqn (S1)–(S3) of the ESI† and the values from Table 1. | ||
When there are two molecules in the asymmetric unit, a doubling of 13C resonances can be observed if the crystallographic environments between otherwise chemically equivalent sites are sufficiently distinct. As shown in Fig. 1b, a 13C chemical shift difference of 2.2 ppm is observed between Cord (δ(13C) = 26.7 ppm) and Cdis (δ(13C) = 24.5 ppm). The 13C signals have been assigned to their sites in the structural model using gauge-including projector augmented-wave (GIPAW)18 DFT calculations as part of CASTEP.19 The calculations were performed for both conformations of Cdis, and the average GIPAW calculated δ(13C) chemical shifts are 26.7 ppm and 24.3 ppm for Cord and Cdis, respectively, resulting in a computed difference of 2.4 ppm. These calculated results, shown on Fig. 1b as sticks, are in excellent agreement with the experimental results.
As the pyrrolidine groups consist of saturated heterocycles, they can exhibit dynamics in the form of ring inversions,20,21 analogous to those observed in cyclohexane.10,22 In order to investigate the dynamics, 1H–13C solid-state CP MAS NMR and T1(13C) measurements were performed at 10 °C steps between 0 °C and 60 °C. As shown in Fig. 1b, there are no significant changes to the 13C chemical shifts of Cord or Cdis as the temperature is increased, and this is also true for all the other resonances (not shown on the figure). Supported by differential scanning calorimetry (see Fig. S5 of the ESI†), this suggests that no phase changes or major structural changes are occurring between these temperatures.
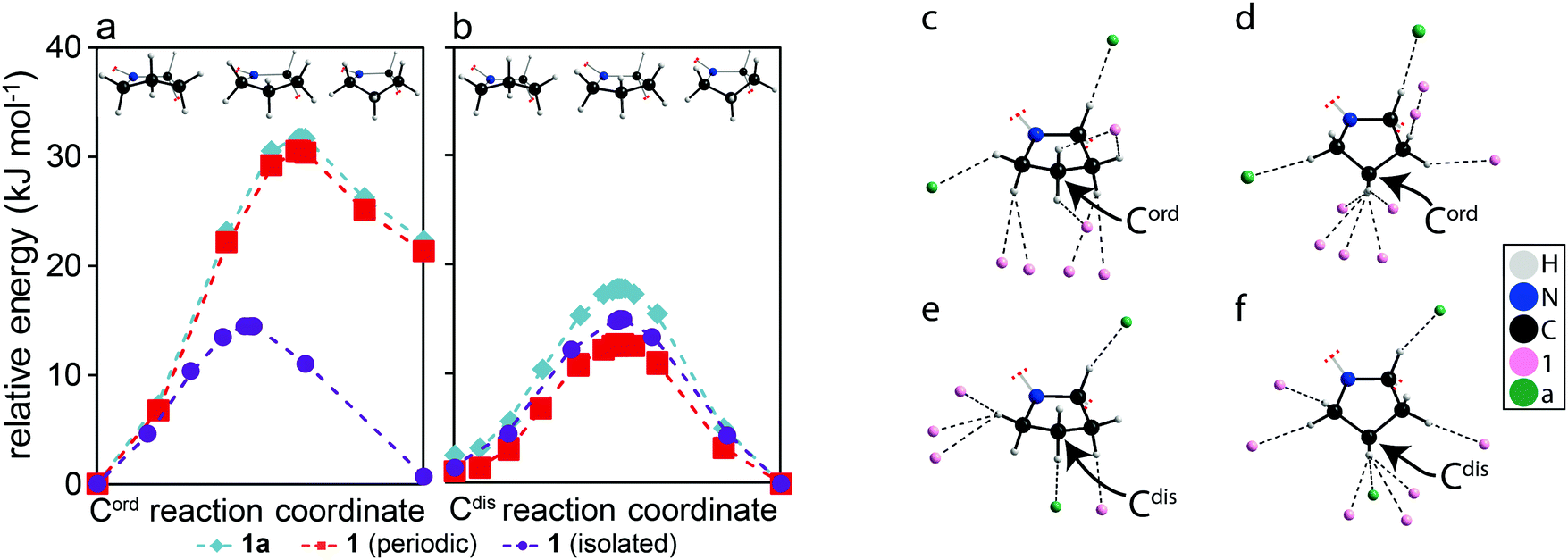 | ||
| Fig. 2 Calculated relative energies as a pyrrolidine group achieves a ring inversion performed on models of (a) Cord and (b) Cdis using: the original structural model of 1a (teal diamonds), 1a maintaining periodicity but with the counterion removed (red squares), and an isolated molecule of 1 (purple circle). A depiction of the models are shown above to provide a frame of reference for the conformations. (c–f) Diagrams showing all atoms within distances shorter than the sum of their van der Waals radius and near hydrogen atoms in either pyrrolidine group of the optimized structural models of 1a when: (c) Cord is puckered up, (d) Cord is puckered down, (e) Cdis is puckered up, (f) Cdis is puckered down. The structure in (d) was obtained from DFT optimizations and has not been experimentally observed. The arrows highlight the carbon atoms of interest. | ||
The T1(13C) at 20 °C for Cord and Cdis were 9.7 s and 1.4 s, respectively, and all T1(13C) values are shown on Fig. 1c and have been tabulated in Table S1 of the ESI.† These short T1(13C) suggest that both Cord and Cdis are dynamic. To place these values into context, the T1(13C) at 20 °C of the rigid carbons of 1a are >100 s, whereas the T1(13C) of rotating methyl groups on 1a are 11 s and 15 s at 20 °C. The relationship between T1(13C), the correlation times (τc), and the activation energy are well known, and have been interpreted using the Bloembergen–Purcell–Pound model.23–25 Assuming it follows the Arrhenius equation, measuring the T1(13C) relaxation times as a function of temperature allows for the activation energy to be extracted (see eqn (S1)–(S3) of the ESI†).
A plot of T1(13C) as a function of the temperature is shown in Fig. 1c, with the fits using eqn (S1)–(S3) (ESI†) being in excellent agreement with the experimental results (Rc2 > 0.96). The extracted activation energies are 11 ± 2 kJ mol−1 for Cord and 16 ± 3 kJ mol−1 for Cdis, with all parameters being summarized in Table 1. The higher activation energy of Cdis is attributed to dynamics in the form of ring inversions, which is also supported by the X-ray structure. In contrast, having a single favourable conformation, short T1(13C) relaxation times, and anisotropic displacement ellipsoids suggesting the presence of vibrations, we associate the lower experimental activation energy of Cord to torsional librations rather than ring inversions. The activation energy of Cdis is very similar in value with the calculated energy of 17.2 kJ mol−1 for the pyrrolidine group in proline performing a ring inversion.26
In order to understand why Cdis is capable of exhibiting ring inversions while Cord is only librating, transition state calculations were performed using CASTEP. The calculations performed on the full structures, shown as teal diamonds in Fig. 2a and b, indicate that the two conformations of the pyrrolidine group, puckered up (left) and down (right), have approximately equal energies for Cdis, whereas a 22.3 kJ mol−1 energy difference is observed for Cord. These results suggest that both conformations of Cdis are energetically favourable, while only the conformation that was experimentally observed in the structural model for Cord is favourable. Further, the calculated transition state energy barrier for Cdis relative to the puckered down conformation is 17.8 kJ mol−1, in excellent agreement with the experimentally measured activation energy of 16 ± 3 kJ mol−1. We propose that this energy barrier is low enough to permit the pyrrolidine ring to undergo dynamics in the form of ring inversions, with both conformations of Cdis being accessible. In contrast, the transition state energy of a ring inversion for Cord is 31.7 kJ mol−1 relative to the starting geometry, which is not in agreement with the experimental NMR results of 11 ± 2 kJ mol−1. The clear discrepancy between the computational and experimental results for Cord suggests that for this ring, the barrier for a ring inversion is too high, and thus libration is observed experimentally.‡ To understand why Cdis is exhibiting ring inversions while Cord is only librating, a series of structural models were created. The first set of models consisted of structure 1a but the counterions “a” have been removed while maintaining periodicity (red squares in Fig. 2a and b), and the second set consisted of completely isolating either molecule of 1 in a cell enlarged by 9 Å along the a, b, and c axes of the unit cell (purple circles in Fig. 2a and b). These models allow interactions arising from the crystal packing to be removed selectively.27
As shown in Fig. 2a and b, there is a clear contribution to the transition state energies from crystal packing. The intermolecular interactions involving Cord and Cdis can be identified based on the molecules involved, either between two molecules of 1 (denoted here as 1⋯1) or between molecules of 1 and a (denoted here as 1⋯a). In the case of Cdis, removing the counterion a has lowered the calculated energy barrier by 5.1 kJ mol−1, whereas a reduction of 2.8 kJ mol−1 was observed upon isolating the molecule 1 that has Cdis. In contrast, isolating the molecule 1 that has Cord reduced the energy barrier of Cord by 17.2 kJ mol−1, whereas removing the counterion a merely reduced the barrier by 1.2 kJ mol−1. Notably, in the isolated molecule of 1, the energy of both conformations of Cord are now nearly the same, and the energy barriers are similar for both Cdis and Cord due to the removal of the intermolecular interactions. Evidently, the intermolecular interactions involving Cord and Cdis are distinct, with 1⋯1 interactions playing a larger role in the energy barrier for Cord and 1⋯a interactions being more important for Cdis.
The interactions in the structural model were analysed in detail and are shown in Fig. 2c–f, illustrating all atoms within distances shorter than the sum of their van der Waals radius and near hydrogen atoms for both conformations (puckered up & down) of both pyrrolidine rings. Cord exhibits mostly 1⋯1 interactions (Fig. 2c and d), whereas Cdis presents both 1⋯1 and 1⋯a interactions (Fig. 2e and f). In the case of Cdis, there are six and eight atoms within this specified radius of any hydrogen atom in the ring when puckered up and down, respectively, compared to eight and ten atoms for Cord in the same conformations. The significance of these interactions was further investigated using DFT calculations performed on molecular cluster models (tabulated in Table S2 of the ESI†).28
The energy barrier for the ring inversions of Cdis appears to originate primarily from the weakening of C–H⋯O interactions originating from 1⋯a, with a difference of 7.4 kJ mol−1 between the puckered up conformation and the transition state, thus destabilizing the transition state of Cdis. This further supports the results obtained from the calculations presented in Fig. 2b, where removing the counterion reduced the energy barrier of Cdis. However, the overall calculated energy barrier (17.8 kJ mol−1) is still small enough to allow ring inversions to occur. In terms of Cord, the destabilizing 1⋯1 interactions originate mainly from a build-up of close contacts between neighbouring pyrrolidine hydrogens (C–H⋯H–C), and in part due to less efficient C–H⋯π interactions in the transition state. Overall, this imposes a much greater energy barrier for a ring inversion to occur (31.7 kJ mol−1), and results in a higher relative energy for the puckered down conformation. This explains why a significant reduction in the energy barrier for Cord was observed in the isolated molecule (cf.Fig. 2a).
In order to investigate the wider significance of the phenomenon investigated here, we have searched the Cambridge Structural Database (in version 5.41)29 for disordered pyrrolidine rings using the structure on Fig. 1a as the search query. Full details on the analysis can be found in Section 4 of the ESI.† We have identified 179 examples where the pyrrolidine ring exhibits structural disorder, with 20 structures having a Z′ = 2 and a case of contrasting disorder akin to our compound 1a (see Table S3 of the ESI†). Further, pyrrolidine ring inversions are shown to have implications on the structure of proline26,30,31 and proline-containing peptides.32,33 Evidently, disorder in pyrrolidine rings is not a rare occurrence, and is likely also in other five-membered rings. Interestingly, while intermolecular interactions have previously been shown to play a role in dynamics,34–36 their influence has been manifested here as two pseudosymmetric pyrrolidine groups exhibiting distinct dynamics.
In conclusion, the disorder observed in Cdis of compound 1a has been attributed to the occurrence of dynamics in the form of ring inversions with an activation energy of 16 ± 3 kJ mol−1. Despite the pseudosymmetry of the structure (Z′ = 2), ring inversions were only observed for Cdis while Cord was constrained to torsional librations with an activation energy of 11 ± 2 kJ mol−1. DFT calculations suggest that the constraints on Cord originate from neighbouring C–H⋯H–C and less effective C–H⋯π intermolecular interactions between 1⋯1 in the transition state and the ring inversion product.
This work was funded by Innovate UK and AstraZeneca (Grant number: KTP11570). We thank Dr G. Whitehead and Dr I. J. Vitorica-Yrezabal at the University of Manchester for X-ray crystallography services, Prof. J. Lewandowski from the University of Warwick for discussions about the NMR relaxation analysis, and Dr R. Storey from AstraZeneca for the DSC results. Data for this study are provided as a supporting data set from WRAP, the Warwick Research Archive Portal at https://wrap.warwick.ac.uk/142932/.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. M. Couillaud, P. Espeau, N. Mignet and Y. Corvis, ChemMedChem, 2019, 14, 8–23 CrossRef CAS.
- R. Censi and P. Di Martino, Molecules, 2015, 20, 18759–18776 CrossRef CAS.
- S. L. Morissette, S. Soukasene, D. Levinson, M. J. Cima and O. Almarsson, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2180–2184 CrossRef CAS.
- E. Pindelska, A. Sokal and W. Kolodziejski, Adv. Drug Delivery Rev., 2017, 117, 111–146 CrossRef CAS.
- G. A. Monti, A. K. Chattah and Y. G. Linck, Annu. Rep. NMR Spectrosc., 2014, 83, 221–269 CrossRef.
- P. C. Vioglio, M. R. Chierotti and R. Gobetto, Adv. Drug Delivery Rev., 2017, 117, 86–110 CrossRef.
- T. Venâncio, L. M. Oliveira, T. Pawlak, J. Ellena, N. Boechat and S. P. Brown, Magn. Reson. Chem., 2019, 57, 200–210 CrossRef.
- D. C. Apperley, A. F. Markwell, I. Frantsuzov, A. J. Ilott, R. K. Harris and P. Hodgkinson, Phys. Chem. Chem. Phys., 2013, 15, 6422–6430 RSC.
- M. Dračínský and P. Hodgkinson, CrystEngComm, 2013, 15, 8705–8712 RSC.
- G. A. Facey, T. J. Connolly, C. Bensimon and T. Durst, Can. J. Chem., 1996, 74, 1844–1851 CrossRef CAS.
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Int. Rev. Phys. Chem., 2017, 36, 39–115 Search PubMed.
- A. S. Tatton, H. Blade, S. P. Brown, P. Hodgkinson, L. P. Hughes, S. O. Nilsson Lill and J. R. Yates, Cryst. Growth Des., 2018, 18, 3339–3351 CrossRef CAS.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45(suppl 1), S174–S186 CrossRef CAS.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS.
- B. Elena, G. Pintacuda, N. Mifsud and L. Emsley, J. Am. Chem. Soc., 2006, 128, 9555–9560 CrossRef CAS.
- C. M. Widdifield, J. D. Farrell, J. C. Cole, J. A. K. Howard and P. Hodgkinson, Chem. Sci., 2020, 11, 2987–2992 RSC.
- C. M. Widdifield, S. O. Nilsson Lill, A. Broo, M. Lindkvist, A. Pettersen, A. Svensk Ankarberg, P. Aldred, S. Schantz and L. Emsley, Phys. Chem. Chem. Phys., 2017, 19, 16650–16661 RSC.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- G. Pfafferott, H. Oberhammer, J. E. Boggs and W. Caminati, J. Am. Chem. Soc., 1985, 107, 2305–2309 CrossRef CAS.
- K. F. Dziubek and A. Katrusiak, Phys. Chem. Chem. Phys., 2011, 13, 15428–15431 RSC.
- F. A. L. Anet and A. J. R. Bourn, J. Am. Chem. Soc., 1967, 89, 760–768 CrossRef CAS.
- D. A. Torchia and A. Szabo, J. Magn. Reson., 1982, 49, 107–121 CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- P. A. Beckmann, Phys. Rep., 1988, 171, 85–128 CrossRef.
- J. Kapitán, V. Baumruk, V. Kopecký Jr, R. Pohl and P. Bour, J. Am. Chem. Soc., 2006, 128, 13451–13462 CrossRef.
- J. R. Yates, T. N. Pham, C. J. Pickard, F. Mauri, A. M. Amado, A. M. Gil and S. P. Brown, J. Am. Chem. Soc., 2005, 127, 10216–10220 CrossRef CAS.
- A. Halme, M. J. Quayle, S. O. Nilsson Lill, A. Pettersen, M. Fransson and C. Boissier, Cryst. Growth Des., 2019, 19, 3670–3680 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- H. S. Park, B. J. Byun, D. Motooka, K. Kawahara, M. Doi, T. Nakazawa, Y. Kobayashi and Y. K. Kang, Biopolymers, 2012, 97, 629–641 CrossRef CAS.
- A. Muralidharan, J. R. Schmidt and A. Yethiraj, J. Phys. Chem. B, 2020, 124, 5899–5906 CrossRef CAS.
- S. K. Sarkar, D. A. Torchia, K. D. Kopple and D. L. VanderHart, J. Am. Chem. Soc., 1984, 106, 3328–3331 CrossRef CAS.
- M. Sonea, H. Yoshimizub, H. Kurosua and I. Ando, J. Mol. Struct., 1993, 301, 227–230 CrossRef.
- P. M. J. Szell, S. Zablotny and D. L. Bryce, Nat. Commun., 2019, 10, 916 CrossRef.
- S. Khazaei and D. Sebastiani, J. Chem. Phys., 2016, 145, 234506 CrossRef.
- J. Baudry, J. Am. Chem. Soc., 2006, 128, 11088–11093 CrossRef CAS.
- J. Claudot, W. J. Kim, A. Dixit, H. Kim, T. Gould, D. Rocca and S. Lebègue, J. Chem. Phys., 2018, 148, 064112 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, equations, and results. See DOI: 10.1039/d0cc05236a |
| ‡ The expected mean absolute error in absolute interaction energies (IE) for PBE-TS calculations is 1.5 kJ mol−1,37 and errors in relative IEs are expected to be even smaller. |
| This journal is © The Royal Society of Chemistry 2020 |