
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect electrochemical synthesis of oxygenates from ethane using phosphate-based electrolysis cells†
Yusuke
Honda
 a,
Naoya
Fujiwara
a,
Shohei
Tada
b,
Yasukazu
Kobayashi
c,
Shigeo Ted
Oyama
de and
Ryuji
Kikuchi
*a
a,
Naoya
Fujiwara
a,
Shohei
Tada
b,
Yasukazu
Kobayashi
c,
Shigeo Ted
Oyama
de and
Ryuji
Kikuchi
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: rkikuchi@chemsys.t.u-tokyo.ac.jp
bDepartment of Materials Sciences and Engineering, Ibaraki University, Ibaraki 316-8511, Japan
cInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), Ibaraki 305-8565, Japan
dCollege of Chemical Engineering, Fuzhou University, Fuzhou 350116, China
eDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA 24061, USA
First published on 28th August 2020
Abstract
Ethane was converted directly to acetaldehyde and ethanol by partial oxidation at 220 °C and ambient pressure using an electrolysis cell with a proton-conducting electrolyte, CsH2PO4/SiP2O7, and Pt/C electrodes. The ethane conversion and the selectivity to the products increased with the voltage applied to the cell. It was found that O species generated by water electrolysis functioned as a favorable oxidant for partial oxidation of ethane on the Pt/C anode at intermediate temperatures. The production rates of acetaldehyde and ethanol recorded in this study were significantly higher than those in preceding reports.
Natural gas from shale has received much attention as a low-cost and abundant energy source. It mainly consists of methane but also contains substantial amounts of ethane. The number of studies on effective ethane utilization has increased steeply in recent years. Most studies have focused on ethylene production,1–6 while few have addressed oxygenate production such as acetaldehyde and ethanol.7–9 These products are difficult to obtain directly from ethane because ethane activation and suppression of over-oxidation of oxygenates are challenging. To overcome these barriers, considerable effort has been made so far focusing on oxidizing species generated in reaction systems. Prior research has confirmed that transient intermediate oxidizing species such as O* and OH* are more effective than molecular oxygen for the selective synthesis of oxygenates. Several researchers have tried thermal catalytic10–13 or electrochemical14,15 approaches to generate transient oxidizing species at various temperatures. The advantages of an electrochemical approach are the controllability of reaction rates by changing the rate of O2− or H+ ion pumping and the availability of inexpensive oxidants (O2,15 CO2,16 and H2O). In the 1990s, Frusteri et al.14 and Hamakawa et al.15 conducted ethane oxidation employing electrochemically-generated O species at around 80 °C and 500 °C, respectively. They successfully synthesized oxygenates by using the electrochemically-generated O species. However, the reaction temperatures greatly influenced the selectivity towards the oxygenates and the ethane conversion: low temperature operation (<100 °C) was insufficient to activate ethane, while the high temperature (>400 °C) promoted over-oxidation of the oxygenates. Accordingly, the increase in the production rates of oxygenates is still a challenging issue.
In this work, we expect that the electrochemical reaction at intermediate temperatures (100–400 °C) can attain high production rates of oxygenates by enhancing both the ethane activation and the suppression of over-oxidation. However, electrochemical ethane oxidation at the intermediate temperatures has not been tested before because there are few appropriate materials for electrolysis cells. Here a composite of CsH2PO4 and SiP2O7 was used as an electrolyte material. The composite showed excellent proton conductivity at around 200 °C under humidified conditions.17 Moreover, the use of oxidizing species (O* and OH*) generated by water electrolysis was targeted for partial oxidation of ethane in the anode (Fig. 1a and b). A commercial Pt/C sheet was employed as the anode because the Pt metal has catalytic activity for the dissociation of C–H bonds in ethane.2 Regarding the utilization of a phosphate-based electrolyte at the intermediate temperatures, most studies have demonstrated its application in fuel cells18–20 and very few studies have demonstrated its application in electrolysis cells for synthesizing variable chemicals such as ammonia.21,22 No research has been reported on the electrochemical conversion of ethane to oxygenates at the intermediate temperatures so far. Herein, we report that ethane was converted directly to acetaldehyde and ethanol with high production rates using oxygen species generated by water electrolysis at 220 °C.
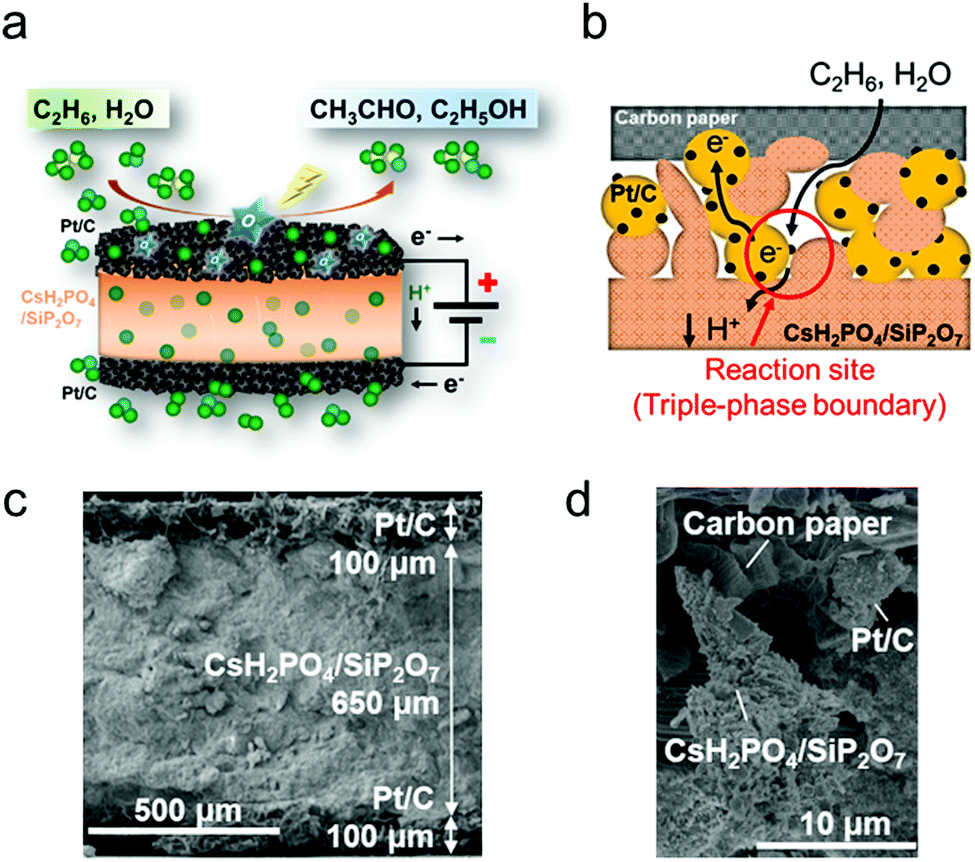 | ||
| Fig. 1 (a) Conceptual diagram of ethane partial oxidation in an electrochemical cell. (b) Schematic illustration of ethane oxidation in the anode. SEM images of (c) the phosphate-based electrolysis cell and (d) the interface between the electrolyte and the Pt/C anode before the reaction. | ||
The construction of the electrochemical cell is explained in the ESI.† Briefly, a mixture of CsH2PO4 and SiP2O7 powder was sandwiched between two commercial Pt/C sheets (10 mm in diameter, Pt loading 1 mg cm−2, where the Pt/C catalyst was loaded on carbon paper) using a uniaxial pelletizer. The XRD patterns for CsH2PO4 and SiP2O7 (Fig. S1, ESI†) show that the two powders were successfully synthesized. Fig. 1c and d show the SEM images of the prepared cell. The thickness of the cell was ∼0.85 mm. The diameter of the secondary particles of Pt/C was ∼5 μm. Rod-shaped electrolyte particles (∼100 nm) and primary Pt/C particles (∼50 nm) were also observed (Fig. S2, ESI†). The SEM image after ethane oxidation (Fig. S3, ESI†) showed that the cross section of the electrolyte composite became dense and the rod structure disappeared. This suggests that the electrolyte material was in a highly viscous molten state at around 200 °C to achieve high H+ conductivity.17 The electrochemical H+ pumping ability of the cell was examined with an I–V test and confirmed to be sufficient (Fig. S4, ESI†). The faradaic efficiency for water splitting was 93% (Table S1, ESI†). The electrochemical partial oxidation of ethane was conducted at 220 °C and ambient pressure using a double-chamber reactor (Fig. S5, ESI†).
Fig. 2a shows the effect of the applied voltage on the ethane conversion and the selectivity to the products (CO2, CO, C2H4, CH3CHO, CH4 and C2H5OH) calculated using eqn (S-1) and (S-2) in the ESI.† The conversion increased from 0.20% to 3.1% with increasing the applied voltage. The selectivity to C2 species such as C2H4, CH3CHO and C2H5OH increased, whereas the selectivity to undesirable CO2 decreased. The production rates of the products are shown in Fig. S6 (ESI†). The production of the C2 species was drastically promoted in the higher voltage region. The current densities at each applied voltage were almost constant during the test as shown in Fig. S7 (ESI†).
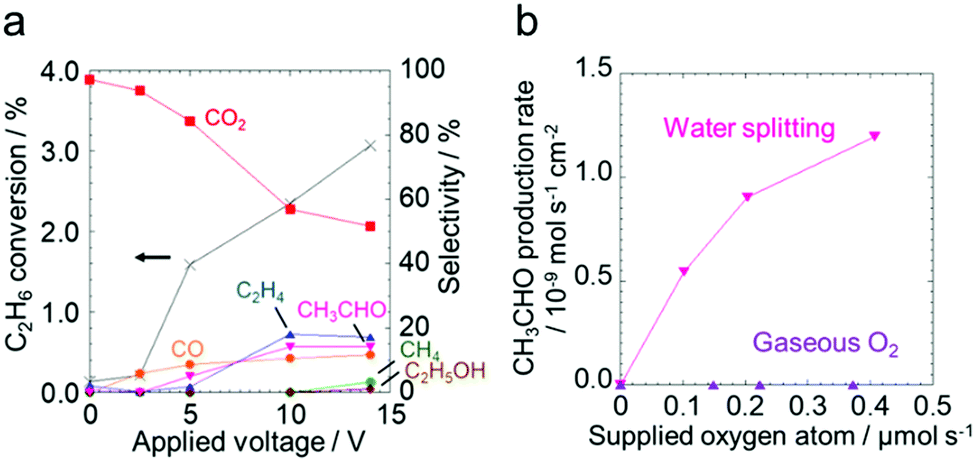 | ||
| Fig. 2 (a) Effect of the applied voltage on the ethane conversion and the product selectivity. (b) Effect of the amount of supplied oxygen atoms generated by water splitting or fed as gaseous O2 on the CH3CHO production rate. Ethane flow rate: 3 mL min−1, temperature: 220 °C, Pt weight: 1 mg cm−2. | ||
Because the electrochemical cell consists of carbon-containing materials such as Pt/C electrodes and the current collectors, there was a possibility for these materials to be sources of the products in the ethane oxidation test. Consequently, blank experiments were conducted to confirm that the products were synthesized from the feed ethane on the Pt/C anode. Table 1 summarizes the production rates of the C2 species under different experimental conditions. In both the Pt/C anode and carbon paper (CP) anode cases, no C2 species were produced when a current density of 50 mA cm−2 was applied without ethane gas (entries 1 and 4). Little amounts of C2H4 were detected under open circuit conditions in an ethane flow (entries 2 and 5). When a current density of 50 mA cm−2 was applied with ethane gas, the Pt/C anode and the CP anode showed a different performance. In the case of the Pt/C anode (entry 3), significant amounts of C2 species were produced. The amounts of produced C2 species, especially C2H4 and CH3CHO, were much larger in the Pt/C anode case (entry 3) than in the CP anode case (entry 6). The main products over the CP anode were CO2 and O2 (entries 4, 6 and Table S5, ESI†). These results indicate that C2H4, C2H5OH and CH3CHO are electrochemically produced in the electrolysis cell from the feed ethane. Moreover, the Pt metal plays an important role in the ethane oxidation to the oxygenates at the intermediate temperatures. The electrochemically-generated O species would react with ethane on the Pt metal at the triple-phase boundary (gas–electrode–electrolyte) as illustrated in Fig. 1b, resulting in the production of the C2 species.
| Entry | Anode | Gas condition (steam, N2) | Current density [mA cm−2] | Production rate [10−9 mol s−1 cm−2] | ||
|---|---|---|---|---|---|---|
| C2H4 | C2H5OH | CH3CHO | ||||
| 1 | Pt/C | Without C2H6 | 50 | 0 | 0 | 0 |
| 2 | With C2H6 | 0 | 0.017 | 0 | 0 | |
| 3 | With C2H6 | 50 | 0.11 | <0.1 | 0.71 | |
| 4 | Carbon paper | Without C2H6 | 50 | 0 | 0 | 0 |
| 5 | With C2H6 | 0 | 0.012 | 0 | 0 | |
| 6 | With C2H6 | 50 | 0.013 | 0 | 0 | |
Fig. 2b shows a comparison of the two types of oxidizing species: gas phase O2 molecules and surface O species derived from water splitting. The CH3CHO production rates are plotted against the number of supplied oxygen atoms. The production rates of the other products are shown in the ESI† (Fig. S8). In addition, the faradaic efficiency for the products is summalized in Tables S2 and S3 (ESI†). The applied voltages were almost constant during the reaction (Fig. S9, ESI†). The production rate of CH3CHO increased with the increase of the oxygen atoms supplied by water electrolysis. On the other hand, gas phase O2 was not consumed to synthesize CH3CHO, but it was involved in the formation of CO2 (Fig. S10, ESI†). This result indicates that CH3CHO was produced by electrochemically-generated O species, and not by gaseous O2. The production rates of the products obtained under different steam partial pressures and at a constant voltage (Fig. S11 and Table S4, ESI†) support the discussion about the oxidizing species. The production rates of the oxygenates increased monotonically with the increase of the current density, indicating that the O species originating from water splitting worked as an effective oxidant. This is consistent with the results shown in Fig. S6, S8, and S11 (ESI†) that once the electrochemically-generated O species was used to form molecular oxygen, the resultant molecular oxygen remained unreacted, and it was consequently detected as one of the products. The dependency of the acetaldehyde production rate on the partial pressure of ethane and the concentration of the O species was analyzed by using the reaction rate equation below (eqn (1)).
| rCH3CHO = k[C2H6]α[O*]β | (1) |
r CH3CHO, k, [C2H6], [O*], α and β indicate the production rate of acetaldehyde, the rate constant, the concentration of ethane, the concentration of electrochemically-generated O species, the kinetic order for ethane and the kinetic order for electrochemically-generated O species, respectively. The reaction test results with different ethane partial pressures are shown in Fig. S12 (ESI†). The kinetic orders, α and β, were determined based on the experimental data (Fig. S11 and S12, ESI†). The details of the assumptions and the calculations are shown in the ESI.† The resultant values of α and β were 0.31 and 1.6, respectively. Based on the value of β which is higher than one (1.6), it is assumed that more than one O atom is involved in the reaction: one O atom is used to abstract the hydrogen atom from ethane and another O atom is inserted into the hydrocarbon fragment. This assumption corresponds with the ethane oxidation steps with the aid of monoatomic oxygen species.15 The experiment without the Pt/C electrode shown in Table 1 (entry 6) demonstrates that the reaction between C2H6 and the O species takes place on the Pt/C electrode not in the gas phase. By considering ethane adsorption on the catalyst surface, the kinetic order for ethane, α, is estimated to be in a range of 0 < α < 1 (details are explained in the ESI†). The obtained kinetic order for ethane (0.31) is in the range, being consistent with the assumption that the absorbed ethane species reacts with the O species.
To investigate the reaction pathway in the anode, electrochemical reactions starting from ethanol or acetaldehyde were conducted. Fig. 3a shows the production rates of the C2 species at different current densities in the ethanol oxidation test. The production rates of the other products are shown in (Fig. S13, ESI†). The production rates of C2H4 and C2H6 increased as the current increased. CH3CHO was produced even under open circuit conditions and its production rate increased upon applying current. It is suggested that a certain amount of CH3CHO is produced from C2H5OH on Pt/C catalytically at an open circuit according to eqn (2).15
| C2H5OH → CH3CHO + H2 | (2) |
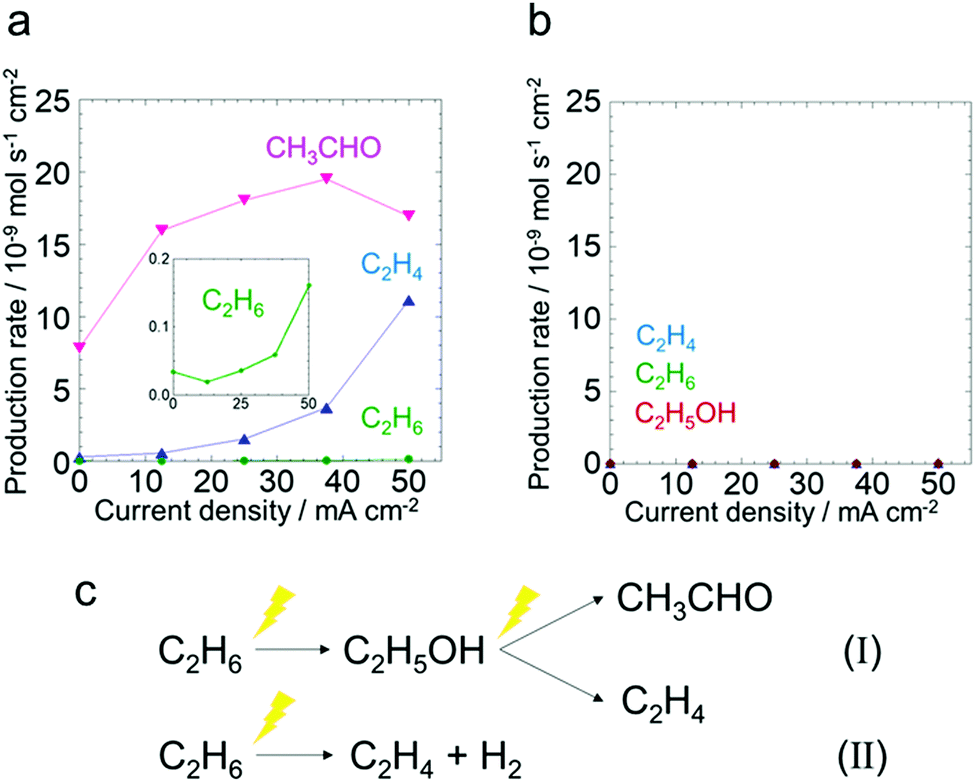 | ||
| Fig. 3 (a) Ethanol oxidation test. The production rates of C2H4, C2H6 and CH3CHO as a function of current density. Ethanol flow rate: 0.8 mL min−1. (b) Acetaldehyde oxidation test. The production rates of C2H4, C2H6 and C2H5OH as a function of current density. Acetaldehyde flow rate: 10 mL min−1, temperature: 220 °C, Pt weight: 1 mg cm−2. (c) Schematic representations of electrochemical ethane oxidation to C2H4, C2H5OH and CH3CHO. | ||
Because H2O was not introduced in the anode in the ethanol oxidation test, H+ production from C2H5OH is a requisite for anodic current generation. Thus, the reactions shown in eqn (3) and (4) might occur, and as a consequence C2H4 and CH3CHO are produced. This indicates that ethanol generated from ethane can be oxidized easily under the anodic polarized conditions.
| C2H5OH → C2H4 + 2H+ + O* + 2e− | (3) |
| C2H5OH → CH3CHO + 2H+ + 2e− | (4) |
Table 2 shows a comparison of the production rates of the C2 species (C2H4, C2H5OH and CH3CHO) obtained with various catalysts in thermal catalytic systems and in electrochemical systems. On a catalyst weight basis, the Pt/C|CsH2PO4/SiP2O7|Pt/C electrolysis cell operated at 220 °C gave much higher production rates of oxygenates and ethylene than those in other studies. The production rates of the other products are summarized as well in Table S6 (ESI†).
| Temperature [°C] | Catalyst | Oxidant | Applied current [mA] | Production rate [μmol g-cat−1 s−1] | Ref. | ||
|---|---|---|---|---|---|---|---|
| C2H4 | C2H5OH | CH3CHO | |||||
| a Electrochemically. b Thermocatalytically. c 30% steam conditions applying 100 mA cm−2. d 40% steam conditions applying 10 V. e Pt containing LTA zeolite. f V2O5 supported on oxidized diamond. | |||||||
| 80a | Nafion-H | O2 | 40 | 0 | 0.26 | 0.54 | 14 |
| 220a | Pt | H2O | 79c | 2.8 | 0 | 1.2 | This work |
| 160d | 5.2 | 0.42 | 16 | ||||
| 475a | Au | O2 | 5.3 | 0 | 0 | 0.16 | 15 |
| 50b | H-LTA-Pte | H2O2 | — | — | 0.0097 | — | 10 |
| 400b | FePO4 | N2O | — | 1.4 | 0.23 | 0.47 | 11 |
| 550b | B/BPO4 | O2 | — | 0.26 | — | 0.84 | 12 |
| 650b | V2O5/O-diaf | CO2 | — | 0.50 | — | 0.039 | 13 |
Partial oxidation of ethane was carried out using a phosphate-based electrolyte and Pt/C electrodes at 220 °C. The ethane conversion and the selectivity to acetaldehyde and ethanol increased under anodically polarized conditions. It was confirmed that C2 species were produced electrochemically from ethane on the Pt/C electrode. Moreover, it was found that O species generated by water electrolysis functioned as an effective oxidant for the synthesis of oxygenates. This report offers for the first time a possibility of ethane conversion to oxygenates at intermediate temperatures using a phosphate-based electrolyte and electrochemically-generated oxidants.
Conflicts of interest
There are no conflicts to declare.References
- A. S. Bodke, D. A. Olschki, L. D. Schmidt and E. Ranzi, Science, 1999, 285, 712–715 CrossRef CAS.
- C. T. Au, K. D. Chen, H. X. Dai, Y. W. Liu and C. F. Ng, Appl. Catal., A, 1999, 177, 185–191 CrossRef CAS.
- D. Melzer, G. Mestl, K. Wanninger, Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 4012 CrossRef.
- P. Sun, G. Siddiqi, W. C. Vining, M. Chi and A. T. Bell, J. Catal., 2011, 282, 165–174 CrossRef CAS.
- N. Mimura, M. Okamoto, H. Yamashita, S. T. Oyama and K. Murata, J. Phys. Chem. B, 2006, 110, 21764–21770 CrossRef CAS.
- Y. Honda, A. Takagaki, R. Kikuchi and S. T. Oyama, Chem. Lett., 2018, 47, 1090–1093 CrossRef CAS.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS.
- R. Jin, M. Peng, A. Li, Y. Deng, Z. Jia, F. Huang, Y. Ling, F. Yang, H. Fu, J. Xie, X. Han, D. Xiao, Z. Jiang, H. Liu and D. Ma, J. Am. Chem. Soc., 2019, 141, 18921–18925 CrossRef CAS.
- J. G. Vitillo, A. Bhan, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2019, 9, 2870–2879 CrossRef CAS.
- B. Liu, S. C. Oh, H. Chen and D. Liu, J. Energy Chem., 2019, 30, 42–48 CrossRef.
- Y. Wang and K. Otsuka, J. Chem. Soc., Faraday Trans., 1995, 91, 3953–3961 RSC.
- Y. Uragami and K. Otsuka, J. Chem. Soc., Faraday Trans., 1992, 88, 3605–3610 RSC.
- K. Okumura, K. Nakagawa, T. Shimamura, N. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Phys. Chem. B, 2003, 107, 13419–13424 CrossRef CAS.
- F. Frusteri, E. N. Savinov, A. Parmaliana, E. R. Savinova, V. N. Parmon and N. Giordano, Catal. Lett., 1994, 27, 355–360 CrossRef CAS.
- S. Hamakawa, K. Sato, T. Hayakawa, A. P. E. York, T. Tsunoda, K. Suzuki, M. Shimizu and K. Takehira, J. Electrochem. Soc., 1997, 144, 1–5 CrossRef CAS.
- Y. Song, L. Lin, W. Feng, X. Zhang, Q. Dong, X. Li, H. Lv, Q. Liu, F. Yang, Z. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 16043–16046 CrossRef CAS.
- S. Yoshimi, T. Matsui, R. Kikuchi and K. Eguchi, J. Power Sources, 2008, 179, 497–503 CrossRef CAS.
- G. Qing, K. Sukegawa, R. Kikuchi, A. Takagaki and S. T. Oyama, J. Appl. Electrochem., 2017, 47, 803–814 CrossRef CAS.
- R. Kikuchi, A. Ogawa, T. Matsuoka, A. Takagaki, T. Sugawara and S. T. Oyama, Solid State Ionics, 2016, 285, 160–164 CrossRef CAS.
- S. M. Haile, C. R. I. Chisholm, K. Sasaki, D. A. Boysen and T. Uda, Faraday Discuss., 2007, 134, 17–39 RSC.
- S. Kishira, G. Qing, S. Suzu, R. Kikuchi, A. Takagaki and S. T. Oyama, Int. J. Hydrogen Energy, 2017, 42, 26843–26854 CrossRef CAS.
- G. Qing, R. Kikuchi, S. Kishira, A. Takagaki, T. Sugawara and S. T. Oyama, J. Electrochem. Soc., 2016, 163, E282–E287 CrossRef CAS.
- S. Liu, K. T. Chuang and J. L. Luo, ACS Catal., 2016, 6, 760–768 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05111j |
| This journal is © The Royal Society of Chemistry 2020 |