
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic oxidative Z/E isomerization of a stilbene favoured by the presence of an electroactive persistent radical†
Paula
Mayorga-Burrezo
 *a,
Christian
Sporer
a,
J. Alejandro
de Sousa
ab,
Nicola
Capra
a,
Klaus
Wurst
c,
Núria
Crivillers
a,
Jaume
Veciana
a and
Concepció
Rovira
*a
*a,
Christian
Sporer
a,
J. Alejandro
de Sousa
ab,
Nicola
Capra
a,
Klaus
Wurst
c,
Núria
Crivillers
a,
Jaume
Veciana
a and
Concepció
Rovira
*a
aDepartment of Molecular Nanoscience and Organic Materials, Institut de Ciència de Materials de Barcelona (ICMAB-CSIC)/CIBER-BBN, Campus Universitari de Bellaterra, E-08193, Cerdanyola, Barcelona, Spain. E-mail: mayorga@vutbr.cz; cun@icmab.es
bLaboratorio de Electroquímica, Departamento de Química, Facultad de Ciencias, Universidad de los Andes, 5101 Mérida, Venezuela
cInstitute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Center for Chemistry and Biomedicine, Innrain 80-82, A-6020 Innsbruck, Austria
First published on 19th October 2020
Abstract
A push–pull-functionalized stilbene has been prepared, with an electroactive perchlorotriphenylmethyl (PTM˙) radical and dimethylamine units as electron-withdrawing and -donating moieties, respectively, showing an electrocatalytic redox-induced Z → E isomerization where the open-shell nature of PTM˙ plays a key role in the isomerization ocurrance.
The rise of molecular machines has entailed a growing interest in the field of molecular switches, thanks to the key role the latter may play in those sophisticated systems.1 Actually, the development of bistable molecules that undergo reversible conversions between their isomers has been a recurrent topic within the scientific community. Among them, particular attention has been devoted to the so-called photoinduced molecular switches,2 based on the light-driven (E/Z) isomerization phenomenon. Several advantages, including high spatiotemporal precision or cleanness,3,4 justify the extensive use of this external stimulus to induce and modulate structural or morphological transitions on the chemical systems. As a result, many are the reports on photoswitchable double bonds that are found in the literature. See for instance: C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (e.g. stilbene),5 NN (e.g., azobenzene),6,7 or CN (e.g. hydrazone).8 However, (E/Z) isomerization reactions of such molecules may require severe photoirradiation conditions,9,10 limiting their use to those applications where intense emission is required.
C (e.g. stilbene),5 NN (e.g., azobenzene),6,7 or CN (e.g. hydrazone).8 However, (E/Z) isomerization reactions of such molecules may require severe photoirradiation conditions,9,10 limiting their use to those applications where intense emission is required.
In a complementary way, it is known that redox reactions can lower the rotation barrier around a double bond by increasing its single-bond character. In fact, electron-transfer-induced (E/Z) isomerizations have been demonstrated in hydrazone11,12 or stilbene derivatives, either through the formation of radical cations13,14 or radical anions.15–17 Furthermore, fast isomerizations can be achieved when catalytic mechanisms involving radical species are activated, as proved for azobenzenes18,19 and stilbene-based structures.20 Consequently, recent advances in molecular machines point at geometric isomerizations triggered by redox reactions as an alternative to photoinduced stimulation.21 For this reason, the design and development of new molecules with suitably substituted double bonds and stable (E/Z) configurations is required. Up to now, to the best of our knowledge, all the studied systems presented electroactive moieties that generate either radical anion or radical cation intermediates upon electron transfer phenomena. Thus, the derived radical charged species were the ones playing a key role on the isomerization process. In this new approach, we address the use of a neutral radical molecule to investigate the impact of the radical character of the starting compound on the isomerization process.
Perchlorotriphenylmethyl (PTM˙) radical unit has proved to be an excellent choice when considering chemical redox switches with optical (linear and nonlinear) and magnetic response.22–24 Some of us recently demonstrated irreversible E → Z isomerizations for two PTM-based stilbene derivatives.25 However, not redox but photo- and thermal-activated processes were investigated in that occasion. Further to this, nothing was said about the radical character since only the hydrogenated (αH-PTM) analogues were considered. On the other hand, some of us also reported the key role that PTM˙ group can play for the stabilization of a vicinal charge defect generated upon oxidation.26 Thus, these previous results motivated us to explore which could be the effect of the electroactive PTM˙ moiety on a redox mediated (E/Z) isomerization. With this aim, a new donor–acceptor-functionalized stilbene dyad, [PTM-DMA]˙ in Scheme 1, with PTM˙ as electron-acceptor was synthesized and characterized. N,N-Dimethylanyline (DMA) was chosen as strong electron-donor moiety, in order to facilitate the oxidation process based on a previous reports on vinylene-bridged PTM-donor derivatives studies with different amine groups.27,28 Additionally, its hydrogenated version (αH-PTM-DMA, see Scheme 1) has also been studied, to gain further insights into the influence of the open-shell character on the phenomenon under study.
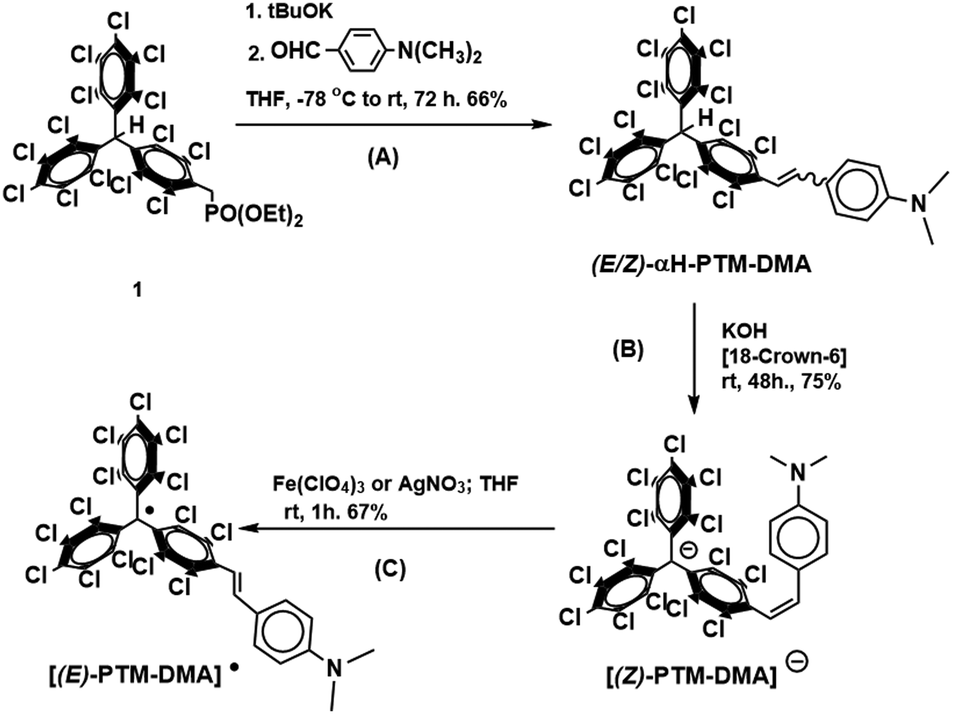 | ||
| Scheme 1 Synthesis of [(E)-PTM-DMA]˙. | ||
The synthetic procedure of [PTM-DMA]˙ started with a Horner–Wadsworth–Emmons coupling (Scheme 1, reaction A) between the phosphonate precursor 129 and 4-dimethylamine-benzaldehyde, yielding αH-PTM-DMA. Remarkably, a different (E/Z) isomeric mixture was obtained depending on the working conditions. According to 1H-NMR monitoring, a 25/75 ratio of E/Z isomers was achieved when the synthesis and the subsequent work-up were performed under daylight conditions (Fig. S1, ESI†). On the contrary, a considerably increase of E/Z ratio (95/5), was achieved under light exclusion (Fig. S2, ESI†). As previously said, these findings were associated with the E → Z photoisomerization (Fig. S3, ESI†) and the subsequently blocked Z → E back conversion previously reported for αH-PTM stilbene compounds.25
Starting from a 15/85 (E/Z)-αH-PTM-DMA mixture, a deprotonation with KOH in presence of [18-crown-6] was then carried out (Scheme 1, reaction B) and, consequently, the corresponding anion ([PTM-DMA]−) was generated. Suitable single crystals of [PTM-DMA]− for X-ray analysis were grown by slow diffusion of hexane into a CH2Cl2 solution of 18-crown-6 K+[PTM-DMA]− (see ESI,† Section S3). The crystal structure unambiguously revealed the formation of the (Z)-isomer; i.e.[(Z)-PTM-DMA]− (Fig. 1A). The solid-state structure of this salt consists in a one-dimensional coordination polymer with the carbanions and their related counterions in alternating positions along the crystallographic a axis (Fig. 1B). Interestingly, a double coordination of each [(Z)-PTM-DMA]− unit with two different K+[18-crown-6] ions was observed.
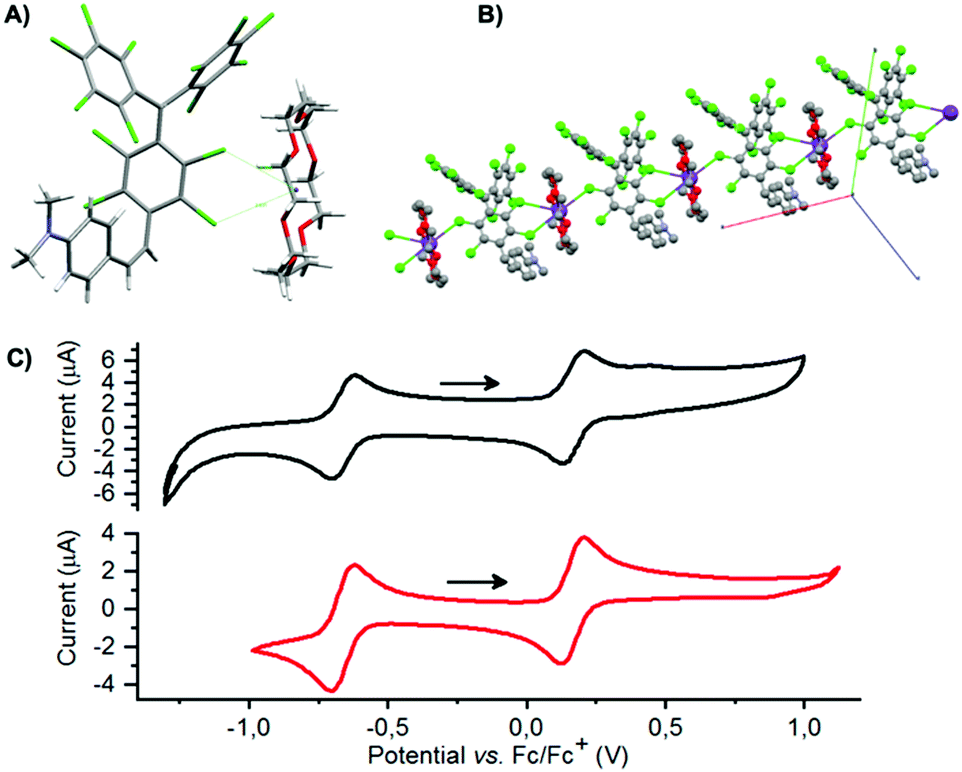 | ||
| Fig. 1 (A) Asymmetric unit in the crystal structure of [(Z)-PTM-DMA]− showing both the anion and the K+[18-crown-6] counterion. Disordered CH2Cl2 molecules have been omitted for clarity. (B) View of the polymeric chain formed along the crystallographic a axis. (C) Cyclic voltammograms (ca. 0.1 mM/0.2 M TBA-PF6 in CH2Cl2) of [(Z)-PTM-DMA]− (black) and [(E)-PTM-DMA]˙ (red) recorded under inert atmosphere at r.t. Scan rate = 0.1 V s−1. WE = Au wire, RE = Ag wire, CE = Pt wire. | ||
Electrochemical properties of the resulting [(Z)-PTM-DMA]− were investigated by cyclic voltammetry (Fig. 1C) and two reversible processes were observed: Ered1/2 = −0.66 V (vs. Fc/Fc+) corresponding to the PTM−/PTM˙ redox couple30 and Eox1/2 = 0.16 V, related to the DMA/DMA˙+ pair.27 According to these data, chemical oxidation of [(Z)-PTM-DMA]− was performed, in order to obtain the same configuration in the [PTM-DMA]˙ radical. Fe(ClO4)3 and AgNO3 were indistinctively used as oxidizing agents (Scheme 1, reaction C). Electron spin resonance (ESR) and UV-Vis electronic absorption spectroscopy fully confirmed the isolation of a radical species (see ESI,† Section S4 and Table S1) upon addition of one equivalent of the oxidant.
The resulting ESR spectrum of the isolated radical shows two main overlapped lines (Fig. S14, ESI†). The two lines are due to the hyperfine coupling of the unpaired spin of the PTM˙ moiety with one proton of the vinylene bridge. The isotropic coupling constant (a(1Ha) = 1.9 G) was in good agreement with those described for different (E)-stilbenoid-based PTM˙ compounds coming from their corresponding (E)-αH-PTM derivatives.29,30 On the other hand, the reddish compound presented the characteristic electronic absorption of conjugated PTM˙ radicals.31 In particular, the features appearing at 392, 495 and 556 nm (Table S1, ESI†) were attributed to the electronic transitions involving the radical frontier orbitals.31 In addition, a low-energy broad band was also detected (λmax = 811 nm, ε ≈ 2875 M−1 cm−1) and assigned to an optically induced electron transfer (ET) from the DMA to PTM˙ unit. TD-DFT calculations (see ESI,† Section S5) supported the assignment. Moreover, previous studies described similar λmax and ε values for ET bands in analogous (E)-isomers of vinylene-bridged PTM-donor dyads27,28,32 generated from their corresponding (E)-αH-PTM precursors. All at once, both the ESR and NIR data suggested the formation of the E isomer of the PTM˙-DMA instead of the Z one. At that point, a different synthetic pathway was carried out for obtaining the [(E)-PTM-DMA]˙ isomer. (E)-αH-PTM-DMA, the (E)-isomer of the precursor, and the intermediate anion [(E)-PTM-DMA]− were considered as starting compounds for the given purpose (see ESI,† Section S2). The resulting radical has identical spectroscopic characteristics than the previous one (Fig. S13, S14 and Table S1, ESI†). Thus, obtaining the thermodynamically more stable [(E)-PTM-DMA]˙ isomer irrespective of the (E/Z) configuration of its given precursor was proved.
Redox properties of [(E)-PTM-DMA]˙ were also studied and compared to the ones described for the [(Z)-PTM-DMA]− anion. As shown in Fig. 1C, the two voltammograms are almost identical, which points to a very efficient (Z) → (E) isomerization upon oxidation of [(Z)-PTM-DMA]−. This observation prompted us to study the anion-to-radical oxidation process in depth. Two different stages could be distinguished from the monitorization, via UV-Vis electronic absorption spectroscopy, of the stepwise oxidation of [(Z)-PTM-DMA]− (Fig. 2A). Up to 1.04 eq. of Fe(ClO4)3, a color change from violet to pale orange was observed. Accordingly, once the characteristic UV-Vis band of [(Z)-PTM-DMA]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 vanished, new UV-Vis features at 387, 489, 556 and 780 nm (Table S1, ESI†) increased through isosbestic points, indicating the generation of an intermediate PTM˙-based species. Then, after the addition of a small excess of oxidizing agent (1.25 eq.), an intensification of the color solution took place, turning from pale orange to orange/reddish. At the same time, the UV-Vis electronic absorbance spectra evolved towards the previously described for [(E)-PTM-DMA]˙. In particular, a remarkable red-shift of the bands and an increase in their related absorbance occurred. Identical results were obtained when using AgNO3 as oxidant or via spectroelectrochemistry (Fig. S16, ESI†). It is known that electronic absorption spectra of stilbenes are generally sensitive to configurational effects. In this line, a red shift in their related UV-Vis features can be seen as long as the extension of π-conjugation within the stilbene chromophore itself and between the stilbene core and unsaturated auxochromic groups increases.20 Thus, this intermediate species was necessarily associated with a less π-conjugated PTM radical-based species than [(E)-PTM-DMA]˙.
33 vanished, new UV-Vis features at 387, 489, 556 and 780 nm (Table S1, ESI†) increased through isosbestic points, indicating the generation of an intermediate PTM˙-based species. Then, after the addition of a small excess of oxidizing agent (1.25 eq.), an intensification of the color solution took place, turning from pale orange to orange/reddish. At the same time, the UV-Vis electronic absorbance spectra evolved towards the previously described for [(E)-PTM-DMA]˙. In particular, a remarkable red-shift of the bands and an increase in their related absorbance occurred. Identical results were obtained when using AgNO3 as oxidant or via spectroelectrochemistry (Fig. S16, ESI†). It is known that electronic absorption spectra of stilbenes are generally sensitive to configurational effects. In this line, a red shift in their related UV-Vis features can be seen as long as the extension of π-conjugation within the stilbene chromophore itself and between the stilbene core and unsaturated auxochromic groups increases.20 Thus, this intermediate species was necessarily associated with a less π-conjugated PTM radical-based species than [(E)-PTM-DMA]˙.
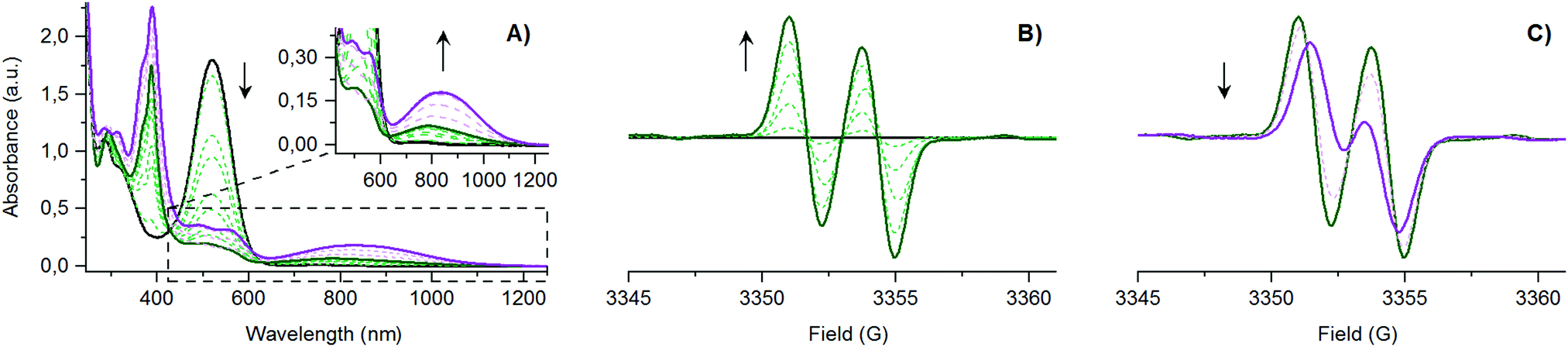 | ||
| Fig. 2 In situ monitorization via: (A) UV-Vis electronic absorption spectroscopy and (B/C) ESR, of the stepwise chemical oxidation of [(Z)-PTM-DMA]− (5 × 10−5 M in THF with Fe(ClO4)3 as oxizing agent (ox.), at r.t., according to Scheme 1, reaction C. Same results were obtained with ox = AgNO3. [(Z)-PTM-DMA]−: solid black line, [(Z)-PTM-DMA]˙: solid green line, and [(E)-PTM-DMA]˙: solid purple line. | ||
In line with UV-Vis results, the two stages of the stepwise chemical oxidation of [(Z)-PTM-DMA]− were also differentiated by in situ ESR. As shown in Fig. 2B, the silent response from the anionic species was firstly transformed into a two-line ESR spectrum when 1.04 eq. of oxidant was added. Interestingly, the obtained a(1Ha) was 2.6 G, exceeding in 0.7 G the one observed in the ESR spectrum of [(E)-PTM-DMA]˙ (Table S1, ESI†). Thus, a higher amount of spin density in the olefinic hydrogen nucleus is counted on the intermediate radical species. As expected, further oxidation (up to 1.25 eq.) resulted in the evolution of the ESR signal towards the one associated with [(E)-PTM-DMA]˙ (Fig. 2C). Bearing all these findings in mind, the generation of the [(Z)-PTM-DMA]˙ isomer radical upon oxidation of [(Z)-PTM-DMA]− anion was concluded. According to Scheme 2, a fast one way Z → E isomerization reaction mediated by cationic species would occur yielding [(E)-PTM-DMA]˙, in good agreement with previous reports in stilbene derivatives.34,35 Moreover, taking into account that only a small excess of oxidant was needed (i.e., 0.25 eq.), an electrocatalytic nature of the oxidative isomerization was considered, as equally described for stilbenes20 or azobenzenes.18,19
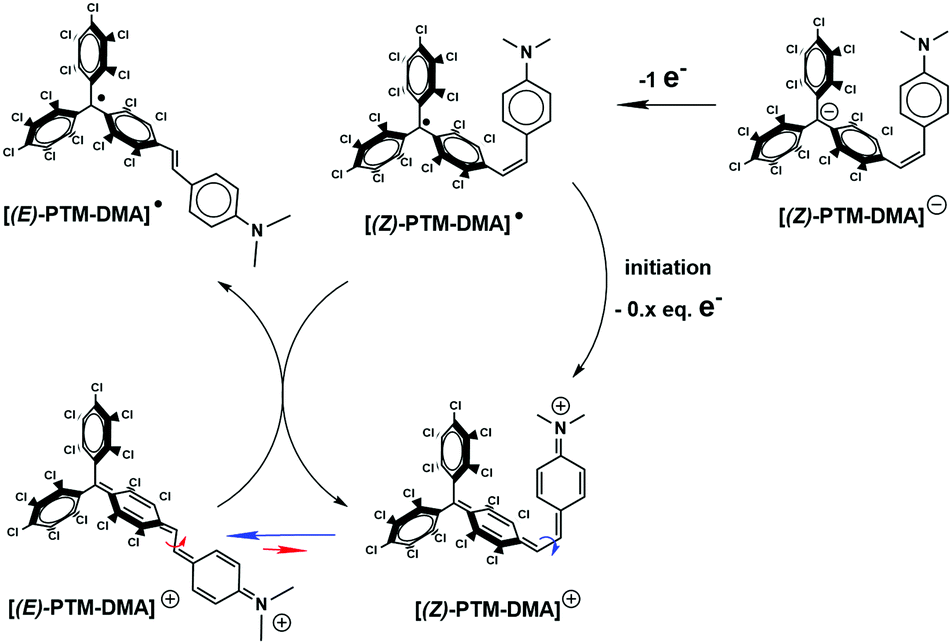 | ||
| Scheme 2 Proposed mechanism for the oxidation process of [(Z)-PTM-DMA]− up to [(E)-PTM-DMA]˙, including the electrocatalytic (i.e., 0.x eq. e−) oxidative (Z) → (E) isomerization occurred through the cationic species [(Z/E)-PTM-DMA]+. | ||
Finally, a high excess of oxidant (>2 eq.) provoked a second color evolution when the orange/reddish solution of [(E)-PTM-DMA]˙ turned into dark blue. Analogously, a new main UV-Vis feature was observed at 600 nm with no hints of ET band (Fig. S18, ESI†). Hence, the generation of the above mentioned closed-shell cationic species, [(E)-PTM-DMA]+, was assumed. The total disappearance of the ESR signal and TD-DFT calculations at a B3LYP/6-31G(d,p) level of theory (Fig. S25 and S26, ESI†) supported the assignment but, unfortunately, this new species was not stable enough to be isolated.
On the contrary, a totally different behavior was found in the case of (Z)-αH-PTM-DMA. The non-radical derivative was subjected to the same chemical oxidation protocol after certifying that the used oxidizing agents have the appropriate redox potentials to form the cation-radical of the amine (Fig. S7, ESI†).36,37 No changes were observed after monitoring by UV-Vis electronic absorption spectroscopy and NMR, demonstrating that the Z → E isomerization does not occur. This result brought to light the need of an open-shell (radical) substituent in the stilbene derivative for the Z → E isomerization process to happen. DFT calculations at the (U)B3LYP/6-31G(d,p) level of theory were run for the transient cationic species of the radical and non-radical derivatives (see ESI,† Section S5). As mentioned, it is known that PTM˙ may contribute to the stabilization of vicinal charge defects generated upon oxidation, by means of a rapid evolution of the structure into a closed-shell quinoidal configuration (Fig. S17, ESI†).26 Accordingly, a lower π bond order at the central olefinic molecular fragment was found for the cation coming from the open-shell dyad (Table S2, ESI†) favouring the isomerization. Similar justification was given to explain the Z/E isomerizations by one-electron oxidation of a few non-radical push–pull-substituted stilbenes.38
In summary, further insights into the redox-induced geometric isomerization phenomenon have been gained from the π-conjugation of an electroactive persistent perchlorotri-phenylmethyl (PTM˙) radical moiety to an E/Z switchable stilbene derivative. An electrocatalytic isomerization has been demonstrated to be undoubtedly triggered by the open-shell nature of PTM˙ unit. We envisage that the findings here reported, in combination with the already described properties (i.e., magnetic, optical or chiral among others) will enhance the prospects of using the PTM˙ unit in smart materials.
This work was supported by the MICIU of Spain (CTQ2016-80030-R and PID2019-111682RB-I00), the Generalitat de Catalunya (2017-SGR-918) and the MINECO (SEV-2015-0496). J. A. de S. is enrolled in the Materials Science PhD program of UAB. J. A. de S. thanks the Spanish Ministry for the FPI fellowship. The authors acknowledge Dr J. Vidal-Galcedo and Dr Vega Lloveras for the ESR measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS.
- J. Zhang, J. Wang and H. Tian, Mater. Horiz., 2014, 1, 169–184 RSC.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- I. Aprahamian, Chem. Commun., 2017, 53, 6674–6684 RSC.
- J. B. Birks, Chem. Phys. Lett., 1976, 38, 437–440 CrossRef CAS.
- G. Orlandi and W. Siebrand, Chem. Phys. Lett., 1975, 30, 352–354 CrossRef CAS.
- I. Cvrtila, H. Fanlo-Virgós, G. Schaeffer, G. Monreal Santiago and S. Otto, J. Am. Chem. Soc., 2017, 139, 12459–12465 CrossRef CAS.
- M. A. Gordillo, M. Soto-Monsalve, C. C. Carmona-Vargas, G. Gutiérrez, R. F. D’vries, J.-M. Lehn and M. N. Chaur, Chem. – Eur. J., 2017, 23, 14872–14882 CrossRef CAS.
- T. Majima, S. Tojo, A. Ishida and S. Takamuku, J. Org. Chem., 1996, 61, 7793–7800 CrossRef CAS.
- T. Majima, S. Tojo, A. Ishida and S. Takamuku, J. Phys. Chem., 1996, 100, 13615–13623 CrossRef CAS.
- I. R. Lee, L. Bañares and A. H. Zewail, J. Am. Chem. Soc., 2008, 130, 6708–6709 CrossRef CAS.
- G. J. Hoijtink and P. H. van der Meij, Zeitschrift für Phys. Chem., 1959, 20, 1–14 CrossRef.
- M. A. Doran and R. Waack, J. Organomet. Chem., 1965, 3, 94–96 CrossRef CAS.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Chem, 2018, 4, 1740–1755 CAS.
- M. Linseis, S. Záliš, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2012, 134, 16671–16692 CrossRef CAS.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS.
- C. Sporer, I. Ratera, D. Ruiz-Molina, J. Vidal Gancedo, K. Wurst, P. Jaitner, C. Rovira and J. Veciana, J. Phys. Chem. Solids, 2004, 65, 753–758 CrossRef CAS.
- C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266–5268 CrossRef CAS.
- I. Ratera, S. Marcen, S. Montant, D. Ruiz Molina, C. Rovira, J. Veciana, J. F. Létard and E. Freysz, Chem. Phys. Lett., 2002, 363, 245–251 CrossRef CAS.
- F. Bejarano, I. Alcon, N. Crivillers, M. Mas-Torrent, S. T. Bromley, J. Veciana and C. Rovira, RSC Adv., 2017, 7, 15278–15283 RSC.
- P. Mayorga Burrezo, C. Franco, R. Caballero, M. Mas-Torrent, F. Langa, J. T. López Navarrete, C. Rovira, J. Veciana and J. Casado, Chem. – Eur. J., 2018, 24, 3776–3783 CrossRef CAS.
- A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2007, 129, 5515–5527 CrossRef CAS.
- A. Heckmann, S. Dümmler, J. Pauli, M. Margraf, J. Köhler, D. Stich, C. Lambert, I. Fischer and U. Resch-Genger, J. Phys. Chem. C, 2009, 113, 20958–20966 CrossRef CAS.
- C. Rovira, D. Ruiz-Molina, O. Elsner, J. Vidal-Gancedo, J. Bonvoisin, J.-P. Launay and J. Veciana, Chem. – Eur. J., 2001, 7, 240–250 CrossRef CAS.
- V. Lloveras, J. Vidal-Gancedo, T. M. Figueira-Duarte, J.-F. Nierengarten, J. J. Novoa, F. Mota, N. Ventosa, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2011, 133, 5818–5833 CrossRef CAS.
- V. Diez-Cabanes, G. Seber, C. Franco, F. Bejarano, N. Crivillers, M. Mas-Torrent, J. Veciana, C. Rovira and J. Cornil, ChemPhysChem, 2018, 19, 2572–2578 CrossRef CAS.
- I. Ratera, C. Sporer, D. Ruiz-Molina, N. Ventosa, J. Baggerman, A. M. Brouwer, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2007, 129, 6117–6129 CAS.
- J. Veciana, J. Riera, J. Castañer and N. Ferrer, J. Organomet. Chem., 1985, 297, 131–141 CrossRef CAS.
- Y. Kuriyama, T. Arai, H. Sakuragi and K. Tokumaru, Chem. Phys. Lett., 1990, 173, 253–256 CrossRef CAS.
- F. D. Lewis, A. M. Bedell, R. E. Dykstra, J. E. Elbert, I. R. Gould and S. Farid, J. Am. Chem. Soc., 1990, 112, 8055–8064 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- B. Kratochvil and R. Long, Anal. Chem., 1970, 42, 43–46 CrossRef CAS.
- H. Spreitzer, M. Scholz, G. Gescheidt and J. Daub, Liebigs Ann., 1996, 1996, 2069–2077 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesic details, experimental procedures and additional spectroscopic and X-ray data. CCDC 1906103. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04973e |
| This journal is © The Royal Society of Chemistry 2020 |