
Selective ring-rearrangement or ring-closing metathesis of bicyclo[3.2.1]octenes†
Evgeniya
Semenova
,
Ouidad
Lahtigui
,
Sarah K.
Scott
,
Matthew
Albritton
,
Khalil A.
Abboud
,
Ion
Ghiviriga
,
Adrian E.
Roitberg
and
Alexander J.
Grenning
 *
*
Department of Chemistry, University of Florida, P. O. Box 117200, Gainesville, FL, USA. E-mail: grenning@ufl.edu
First published on 11th September 2020
Abstract
Explored was the competitive ring-closing metathesis vs. ring-rearrangement metathesis of bicyclo[3.2.1]octenes prepared by a simple and convergent synthesis from bicyclic alkylidenemalono-nitriles and allylic electrophiles. It was uncovered that ring-closing metathesis occurs exclusively on the tetraene-variant, yielding unique, stereochemically and functionally rich polycyclic bridged frameworks, whereas the reduced version (a triene) undergoes ring-rearrangement metathesis to 5–6–5 fused ring systems resembling the isoryanodane core.
3,3-Dicyano-1,5-dienes are attractive substrates due to their ease of construction from ketones, malononitrile, and allylic electrophiles, and their ability to undergo Cope rearrangement.1 In fact, 3,3-dicyano-1,5-dienes are the classic Cope rearrangement substrates.2 We have been studying this class of 1,5-dienes as substrates for complex polycycloalkane synthesis.3–7
We became interested in iterating the deconjugative alkylation8–10 and the diastereoselective[3,3]sigmatropic rearrangement steps of alkylidenemalononitrile functionalization. If performed on alkylidenemalononitriles 1 with allylic electrophiles 2/2′, unique tetraenes 6 could be rapidly established via intermediates 3–5 (Scheme 1). Further piquing our interest was how these substrates would react under olefin metathesis conditions: would they undergo ring-closing metathesis (RCM11) (to 7) or ring-rearrangement metathesis (RRM12) (to 8) (Scheme 2A)? ring-rearrangement metathesis would occur by the ring-opening of the central, strained cycloheptene olefin followed by double ring-closing metathesis, whereas ring-closing metathesis would result simply from the “allylic arms” reacting with each other directly. Notably, the ring-closing route would require the “allyl arms” to be cis-oriented13–16 as well as a conformationally biased17 for the axial isomer. In addition to the growing body of work on the synthesis of bridged bicyclic systems via RCM,13–16 we have previously shown one example of an RCM reaction on a scaffold related to those of interest to this work,6 though there are no possibilities for RRM with this particular substrate (Scheme 2B). Conversely, bridged bicyclo[3.2.1]octenes can react by ring-opening cross-metathesis (ROCM) or polymerization (ROMP) (Scheme 2C).18–21 For example, ketone (Y = O), alcohol (Y = H/OH), and alkylidenemalononitrile-containing scaffolds react via ROCM (Scheme 2C). Thus, we hypothesize that either pathway (RCM or RRM) is plausible and potentially in competition for the proposed scaffolds 6. As such, we began a campaign to explore the reactivity and selectivity of these types of substrates in RCM vs. RRM reactivity. Herein we report that such tetraenes 6 undergo exclusive ring-closing metathesis to 7. We also provide a hypothesis for the observed chemoselectivity, which ultimately yields a method to achieve exclusive ring-rearrangement metathesis to 5–6–5 tricyclic ring systems.
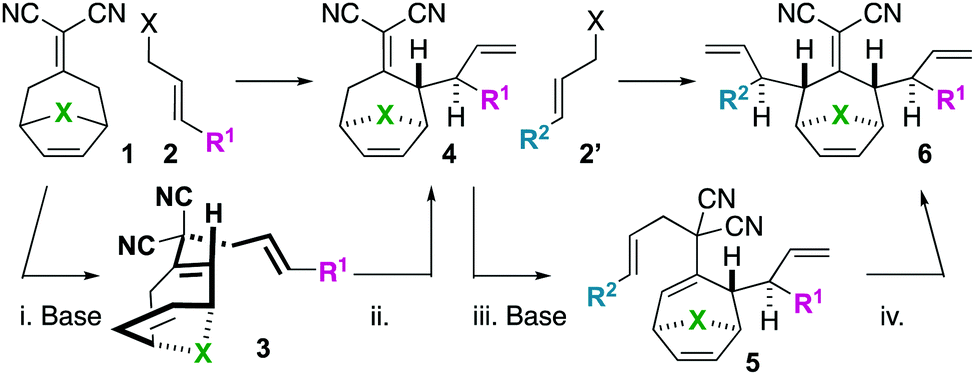 | ||
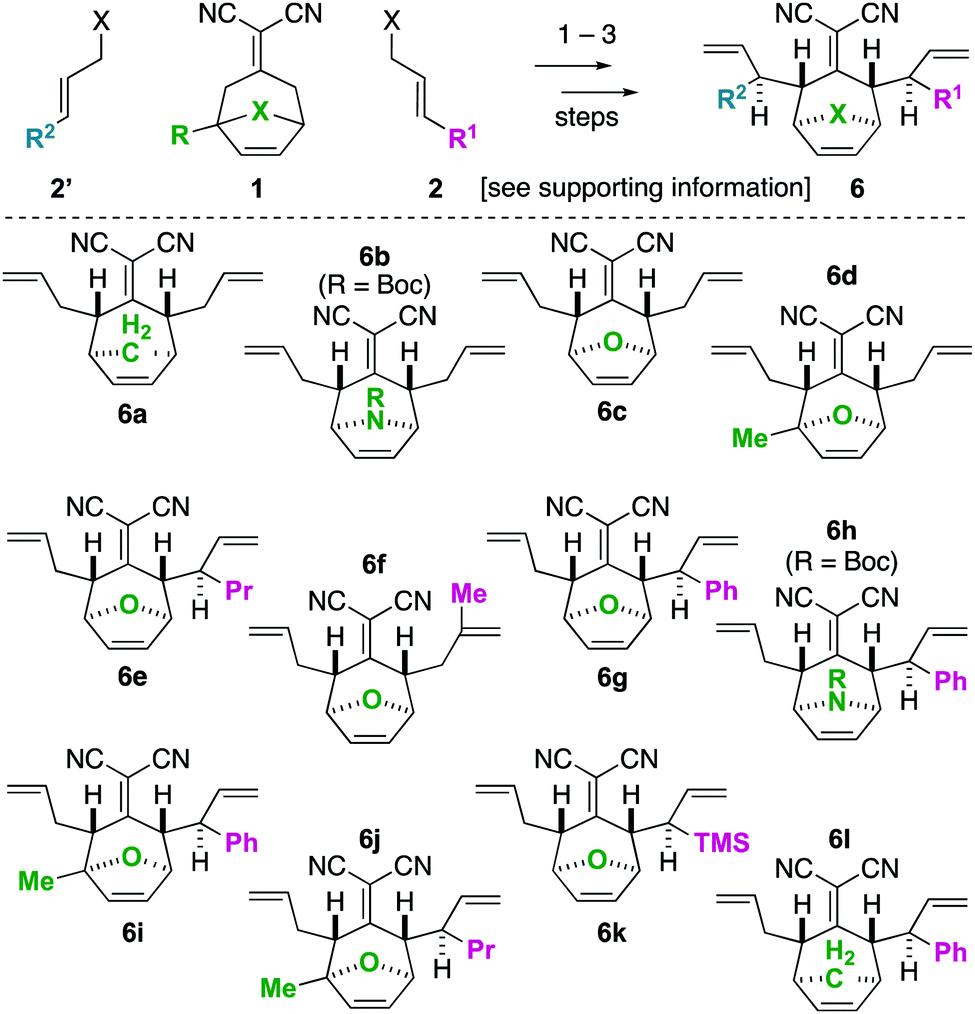
| Scheme 1 Iterative deconjugative allylation/Cope rearrangement to synthesize bicyclic tetraenes (6). | ||
To begin our studies, we prepared tetraenes 6a–6l by iterative deconjugative allylic alkylation/Cope rearrangement (Scheme 3). Depending on the substitution pattern, products 6 are available in 1–3 steps as single diastereomers via diastereoselective Cope rearrangements (see the ESI†). We next turned to the examination of the ring-closing metathesis (RCM) vs. ring-rearrangement metathesis (RRM) question posed for these substrates (Scheme 4). In these studies, standard Ru-based metathesis catalysts (Grubbs-II22 (G-II) or Hoveyda–Grubbs-II22 (HG-II)) were utilized. Substrates 6a–6l underwent clean ring closing metathesis to 7a–7l exclusively under conventional conditions (e.g. nonpolar solvent, r.t. −80 °C, with or without ethylene). Generally speaking, both catalysts examined performed reasonably well. However, in a few side-by-side comparisons, the HG-II catalyst did outperform the G-II catalyst. The first three products in the table (7a–7c) were either cyclopentadiene-(7a), N-Boc-pyrrole-(7b), or furan-(7c) derived. For these substrates, the methylene or heteroatom “X-group” had little to no effect on the efficiency of the transformation. The remaining substrates 6d–6l showcase a variety of substitution patterns and functional groups that were tolerated in the ring-closing metathesis reaction yielding 7d–7l. We also found that the 1,3-diphenylallyl moiety on 6m performed well in the metathesis reaction to yield 7g (Scheme 4). As a final note, preliminary data supports that the sequence can be telescoped: from 5l, the Cope rearrangement and the ring-closing metathesis steps can be performed in one-pot fashion to yield 7e.
 | (1) |
 | (2) |
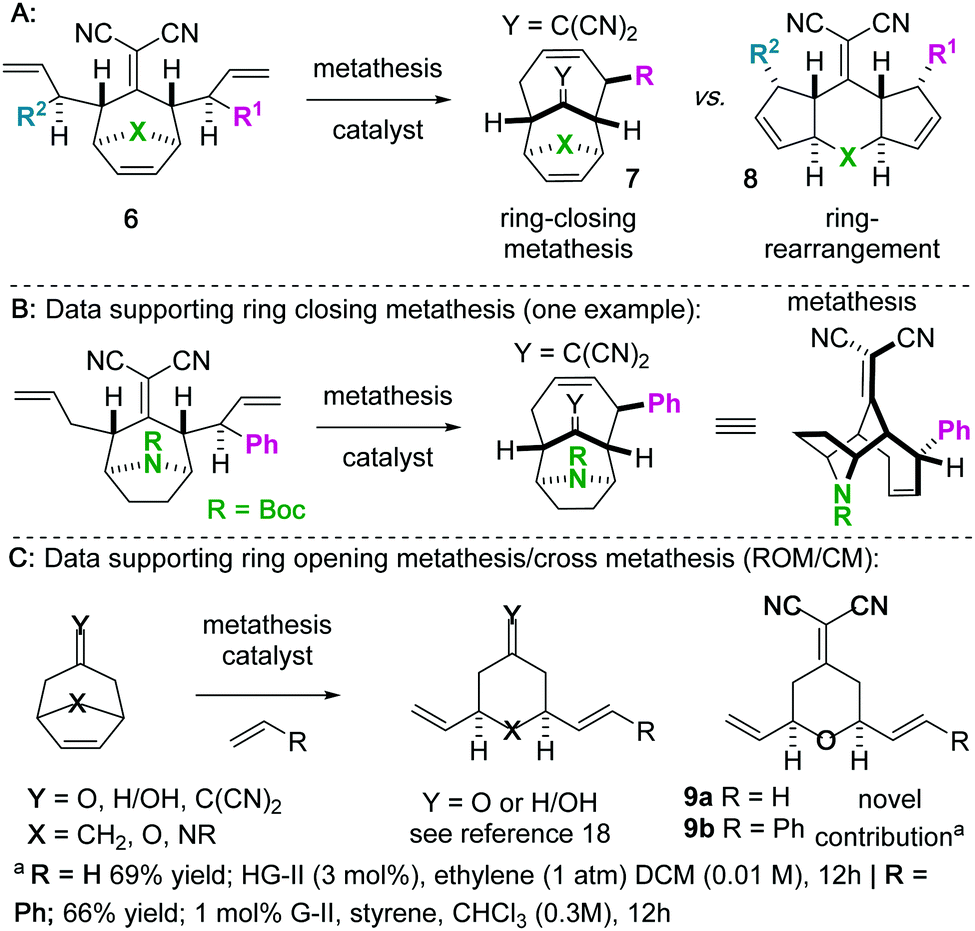 | ||
| Scheme 2 (A) Will bicyclic tetraenes 6 undergo RCM or RRM? (B–C) Support for RCM and RRM. | ||
 | ||
| Scheme 3 Synthesis of bicyclic tetraenes. | ||
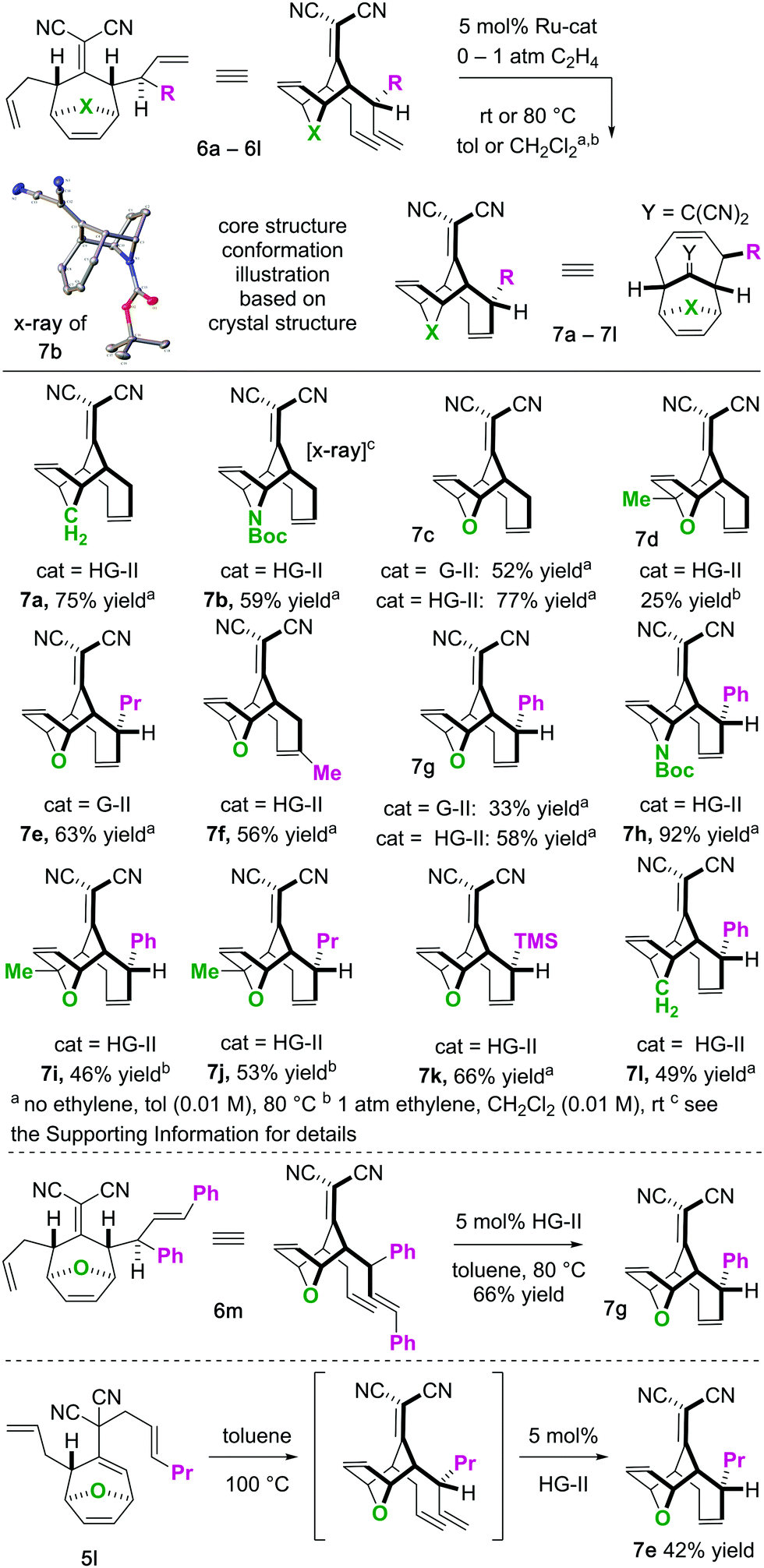 | ||
| Scheme 4 Scope of ring-closing metathesis reaction. | ||
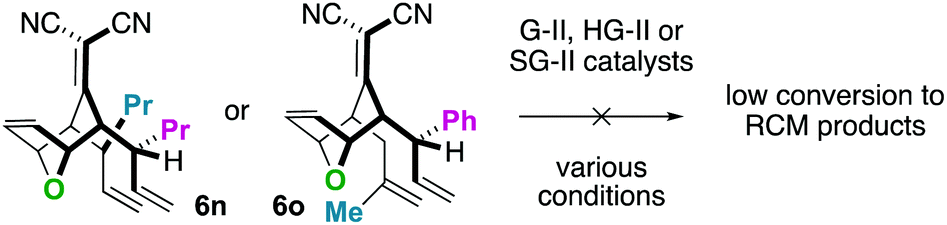
For the substrates in Scheme 4 only one of the “allylic arms” is decorated with an additional substituent. That is because it is generally challenging to perform ring-closing metathesis on densely substituted alkenes. For example, substrates 6n and 6o underwent sluggish and low yielding ring-closing metathesis, even with the Stewart–Grubbs (SG-II; CAS#:[927429-61-6]23) catalyst, which is commonly more accepting to steric challenges (eqn (1)).
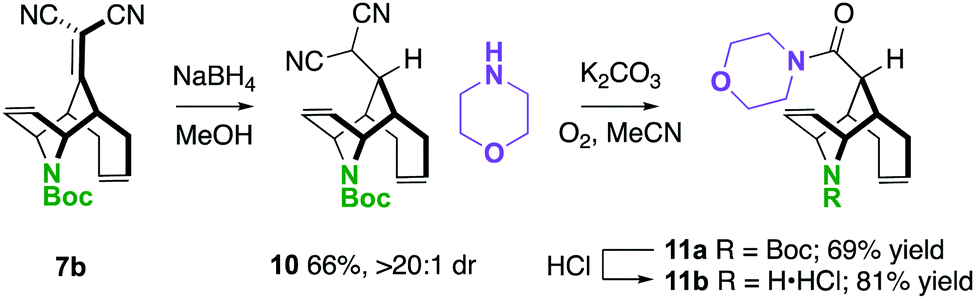
One way in which we envisaged finding potential application of these molecules is described in Scheme 5. In two steps, a unique piperidine carboxamide 11a was prepared by NaBH4 conjugate reduction and oxidative amidation.24 The N-Boc-piperidine can be deprotected to the amine·HCl 11b under standard conditions resulting in an interesting scaffold for drug discovery, considering that they are rigid piperidine carboxylate scaffolds.25
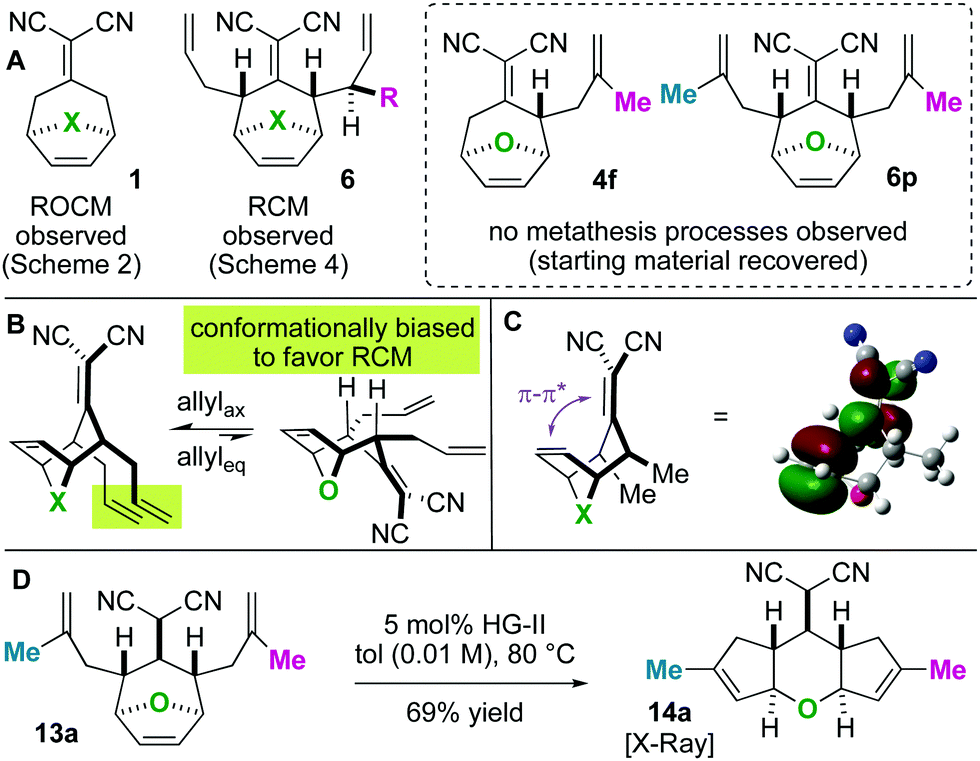 | ||
| Scheme 5 (A) Summary of RCM vs. RRM selectivity. (B and C) Rationale for RCM regioselectivity: conformational bias (B) and an anchimeric effect (C). (D) Alkylidene reduction results in a scaffold that undergoes ring-rearrangement metathesis. | ||
Next, we wished to understand and overturn the observed chemoselectivity for ring-closing metathesis over ring-rearrangement metathesis (Scheme 5). To compare and summarize, non-allylated (1) and bis-allylated (6) scaffolds have unique reactivity to metathesis catalysts: scaffolds 1 undergo ring-opening cross metathesis (ROCM) whereas the bis-allylated variants undergo ring-closing metathesis (RCM). A thought-provoking observation was that 6p was wholly unreactive to metathesis catalysts. Regarding 6p, we presumed that ring-closing metathesis to yield a tetrasubstituted olefin would be unfavourable and therefore ring-rearrangement metathesis would be the dominant pathway. However, no metathesis processes were observed; the starting material was recovered in high yield. Furthermore, even the mono 2-methylallylated scaffold 4f was completely unreactive (starting material recovered). These observations allow us to draw conclusions on the ring-closing vs. ring-rearrangement metathesis chemoselectivity (Scheme 5B and C). First, ring-closing metathesis is favoured when either di- or tri-substituted cyclic olefins are expected. The structures are also conformationally biased, where the “allylic arms” are in an axial-position and thus in close proximity to one another (Scheme 5B). This was confirmed by NMR studies (see the ESI†). And second, as shown in Scheme 5C, we have found an anchimeric effect between the cyclic alkene and the alkylidenemalononitrile (a π–π* interaction). This was achieved computationally where structures were optimized using the DFT level of theory M062x/cc-pvdz.26 Notably, the qualitative trends do not change when using a different functional or basis set combination. We found the localized bond orbitals for the π and π* orbitals of interest, and used the second order energy between them as computed in NBO 3.1 to quantify the extent of the interaction. Specifically, a 1.9 kcal mol−1 interaction energy was found. To remove this through space interaction (anchimeric effect), the alkylidenemalononitrile was reduced yielding 13a, which exclusively underwent ring-rearrangement metathesis to 14a (Scheme 5D).
Having found that ring-rearrangement metathesis can be favoured by alkylidenemalononitrile reduction, we next examined the scope of the RRM transformation (Scheme 6). It was found that a variety of scaffolds with 2-alkylation on the “allylic arms” were competent substrates for ring-rearrangement (14a–c). We expected these substrates to be successful because ring-closing metathesis is prohibited by the substitution patterns on the alkenes. We were pleased to also find that substrates with unsubstituted “allylic arms” also yielded the desired ring-rearrangement metathesis products 14d–14f over the ring-closing metathesis products. It was also observed that 14e could be prepared either from a linear precursor 13e or a cyclic one 10, prepared independently via the chemistry described in Scheme 6. Thus, there are two potential entry routes into the ring-rearranged 5–6–5 scaffolds. As a final result, the malononitrile functional group can be interconverted to amides by Hayashi's oxidative amidation protocol as shown in the conversion of 14d to 15.
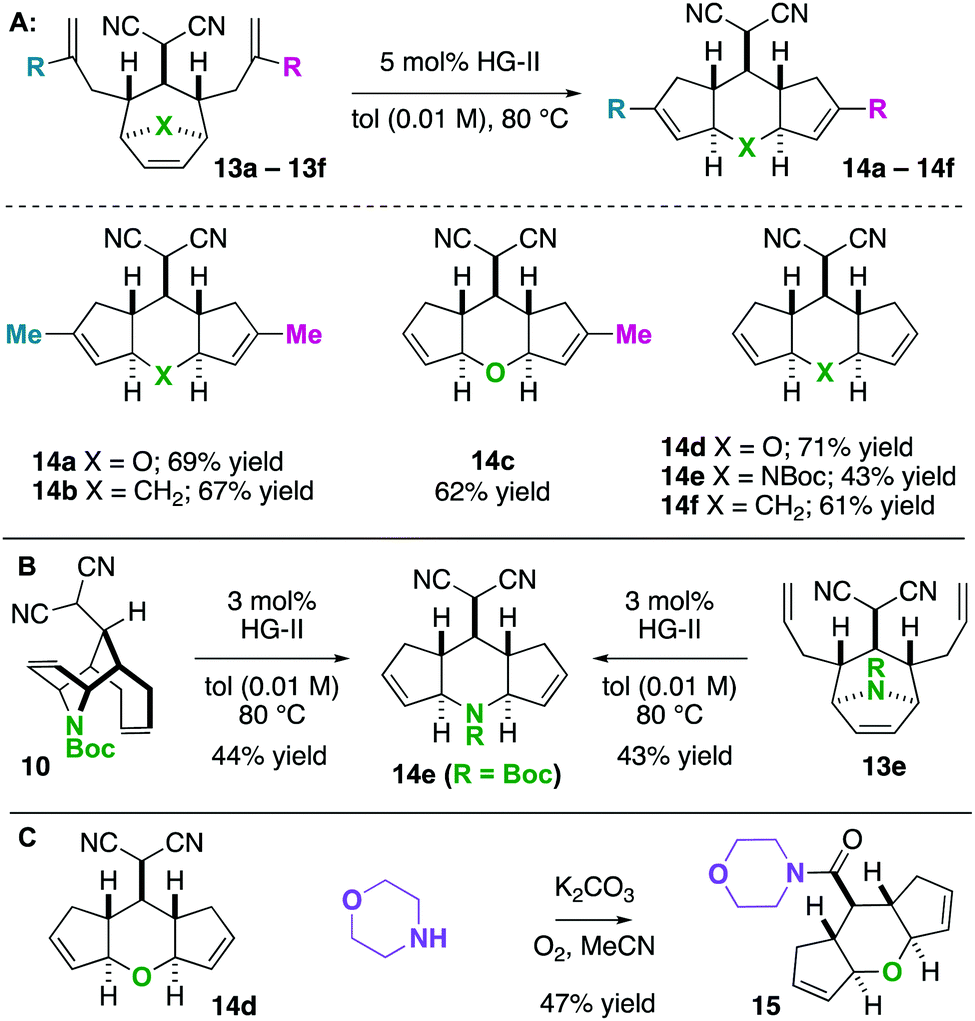 | ||
| Scheme 6 (A and B) Ring rearrangement metathesis to 5–6–5 scaffolds. (C) Oxidative amidation. | ||
We have developed a protocol to assemble bicyclic[3.2.1]tetraenes and explored their reactivity as metathesis substrates. It was uncovered that the tetraenes are kinetically predisposed to undergo ring closing metathesis yielding doubly bridged cyclodeca-1,6-dienes. It was also hypothesized that a π–π* interaction between the strained endocyclic olefin and the alkylidenemalononitrile precluded ring-opening metathesis. In support of this hypothesis, alkylidenemalononitrile reduction can result in chemoselectivity favouring ring-rearrangement metathesis. For both of the scaffolds, the malononitrile functional group can be converted to amides by oxidative amidation. Future studies will involve the exploration of transformations favouring ring-rearrangement metathesis, as this yields frameworks common to natural products such as pepluanone,27,28 retigeranic acid,29 perseanol,30 and leucosceptroids,31–33 among others.
This material is based upon work supported by the National Science Foundation under Grant No. 1844443. We thank the College of Liberal Arts and Sciences and the Department of Chemistry at the University of Florida for start-up funds. We thank Umicore for the generous donation of metathesis catalysts. We thank the Mass Spectrometry Research and Education Center and their funding source: NIH S10 OD021758-01A1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133–3148 RSC.
- A. C. Cope and K. E. Hoyle, J. Am. Chem. Soc., 1941, 63, 733–736 CrossRef.
- F. Emmetiere and A. J. Grenning, Org. Lett., 2020, 22, 842–847 CrossRef.
- P. Vertesaljai, R. Serrano, M. D. Mannchen, M. Williams, E. Semenova and A. J. Grenning, Org. Lett., 2019, 21, 5704–5707 CrossRef.
- E. Fereyduni, J. N. Sanders, G. Gonzalez, K. N. Houk and A. J. Grenning, Chem. Sci., 2018, 9, 8760–8764 RSC.
- E. Fereyduni and A. J. Grenning, Org. Lett., 2017, 19, 4130–4133 CrossRef.
- O. Lahtigui, F. Emmetiere, W. Zhang, L. Jirmo, S. Toledo-Roy, J. C. Hershberger, J. M. Macho and A. J. Grenning, Angew. Chem., Int. Ed., 2016, 55, 15792–15796 CrossRef.
- H. Nakamura, H. Iwama, M. Ito and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 10850–10851 CrossRef.
- R. B. Grossman and M. A. Varner, J. Org. Chem., 1997, 62, 5235–5237 CrossRef.
- P. Vertesaljai, P. V. Navaratne and A. J. Grenning, Angew. Chem., Int. Ed., 2016, 55, 317–320 CrossRef.
- M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073–2077 CrossRef.
- S. Kotha, M. Meshram, P. Khedkar, S. Banerjee and D. Deodhar, Beilstein J. Org. Chem., 2015, 11, 1833–1864 CrossRef.
- A. Morehead Jr. and R. Grubbs, Chem. Commun., 1998, 275–276 Search PubMed.
- C. E. Neipp and S. F. Martin, J. Org. Chem., 2003, 68, 8867–8878 CrossRef.
- A. Michaut, S. Miranda-Garcia, J. C. Menendez and J. Rodriguez, Org. Lett., 2004, 6, 3075–3078 CrossRef.
- M. Lin, P.-J. Cai, Z. Zeng, N. Lin, Y. Shen, B. Tang, F. Li, C. Chen, Z.-X. Yu and Y. Zhang, Chem. – Eur. J., 2018, 24, 2334–2338 CrossRef.
- R. Chen, Y. Shen, S. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14198–14210 CrossRef.
- D. L. Wright, L. C. Usher and M. Estrella-Jimenez, Org. Lett., 2001, 3, 4275–4277 CrossRef.
- M. D. Mihovilovic, B. Groetzl, W. Kandioller, R. Snajdrova, A. Muskotal, D. A. Bianchi and P. Stanetty, Adv. Synth. Catal., 2006, 348, 463–470 CrossRef.
- J. Mandel, N. Dubois, M. Neuburger and N. Blanchard, Chem. Commun., 2011, 47, 10284–10286 RSC.
- P. Disetti, L. Piras, M. Moccia, M. Saviano and M. F. A. Adamo, Eur. J. Org. Chem., 2017, 6202–6208 CrossRef.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef.
- I. C. Stewart, T. Ung, A. A. Pletnev, J. M. Berlin, R. H. Grubbs and Y. Schrodi, Org. Lett., 2007, 9, 1589–1592 CrossRef.
- J. Li, M. J. Lear and Y. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 9060–9064 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- See the ESI† for details related to this computational analysis.
- G. Corea, E. Fattorusso, V. Lanzotti, P. Di Meglio, P. Maffia, G. Grassia, A. Ialenti and A. Ianaro, J. Med. Chem., 2005, 48, 7055–7062 CrossRef CAS.
- E. Barile, E. Fattorusso, A. Ialenti, A. Ianaro and V. Lanzotti, Bioorg. Med. Chem. Lett., 2007, 17, 4196–4200 CrossRef CAS.
- M. Kaneda, R. Takahashi, Y. Iitaka and S. Shibata, Tetrahedron Lett., 1972, 13, 4609–4611 CrossRef.
- A. Han, Y. Tao and S. E. Reisman, Nature, 2019, 573, 563–567 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807–3810 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, Angew. Chem., Int. Ed., 2014, 53, 11351–11355 CrossRef CAS.
- S. Guo, J. Liu and D. Ma, Angew. Chem., Int. Ed., 2015, 54, 1298–1301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1962315. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04624h |
| This journal is © The Royal Society of Chemistry 2020 |