
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum-chemistry-aided ligand engineering for potential molecular switches: changing barriers to tune excited state lifetimes†
Dorottya
Sárosiné Szemes
 a,
Tamás
Keszthelyi
a,
Mariann
Papp
a,
László
Varga
b and
György
Vankó
*a
a,
Tamás
Keszthelyi
a,
Mariann
Papp
a,
László
Varga
b and
György
Vankó
*a
aWigner Research Centre for Physics, P.O. Box 49, Budapest, H-1525, Hungary. E-mail: vanko.gyorgy@wigner.hu
bEvoBlocks Ltd., Berlini u. 47-49., Budapest, H-1045, Hungary
First published on 31st August 2020
Abstract
Substitution of terpyridine at the 4′ position with electron withdrawing and donating groups is used to tune the quintet lifetime of its iron(II) complex. DFT calculations suggest that the energy barrier between the quintet and singlet states can be altered significantly upon substitution, inducing a large variation of the lifetime of the photoexcited quintet state. This prediction was experimentally verified by transient optical absorption spectroscopy and good agreement with the trend expected from the calculations was found. This demonstrates that the potential energy landscape can indeed be rationally tailored by relevant modifications based on DFT predictions. This result should pave the way to advancing efficient theory-based ligand engineering of functional molecules to a wide range of applications.
Molecular systems based on transition metals have long been studied to provide us with possible molecular devices, including switches.1 A great number of Fe(II) complexes show electronic bistability, and at low temperatures back and forth switching with light has been observed between their singlet ground state and metastable quintet excited state.2,3 Exploiting this behavior in devices applicable close to room temperature still seems a long way, as it requires a substantial enhancement of the stability of the photoinduced quintet state.4 This can possibly be achieved by redesigning the potential energy surfaces so that the barrier between the quintet and the singlet state is heightened.
Electron withdrawing (EW) and donating (ED) substituents are tools to reshape the molecular electron density, often utilized to modify positional reactivity in organic chemistry, the behaviour of transition metal complexes for photocatalysis, light-harvesting, luminescence, or spin crossover.5,6 Recent studies revealed how ED/EW substituents change the electronic structure of (pyridine-bearing) ligands in spin-transition systems, shifting the spin-state equilibrium, or the Fe(II) singlet-quintet energy gap.7–9
By judicious selection substituents can be powerful tools to tailor the potential energy surfaces and thereby optimize functionality, and this is exploited in the present study where we apply ligand substitution to systematically modify the quintet lifetime by changing the relevant energy barriers. Halcrow and colleagues demonstrated very substantial substituent effects for γ-substituted pyridin,9 so we have selected several EW/ED groups to substitute the tridentate terpy (2,2′:6′,2′′-terpyridine) ligand in the 4′ position, to study the corresponding effects in their homoleptic Fe(II) complexes (see the inset of Fig. 1).
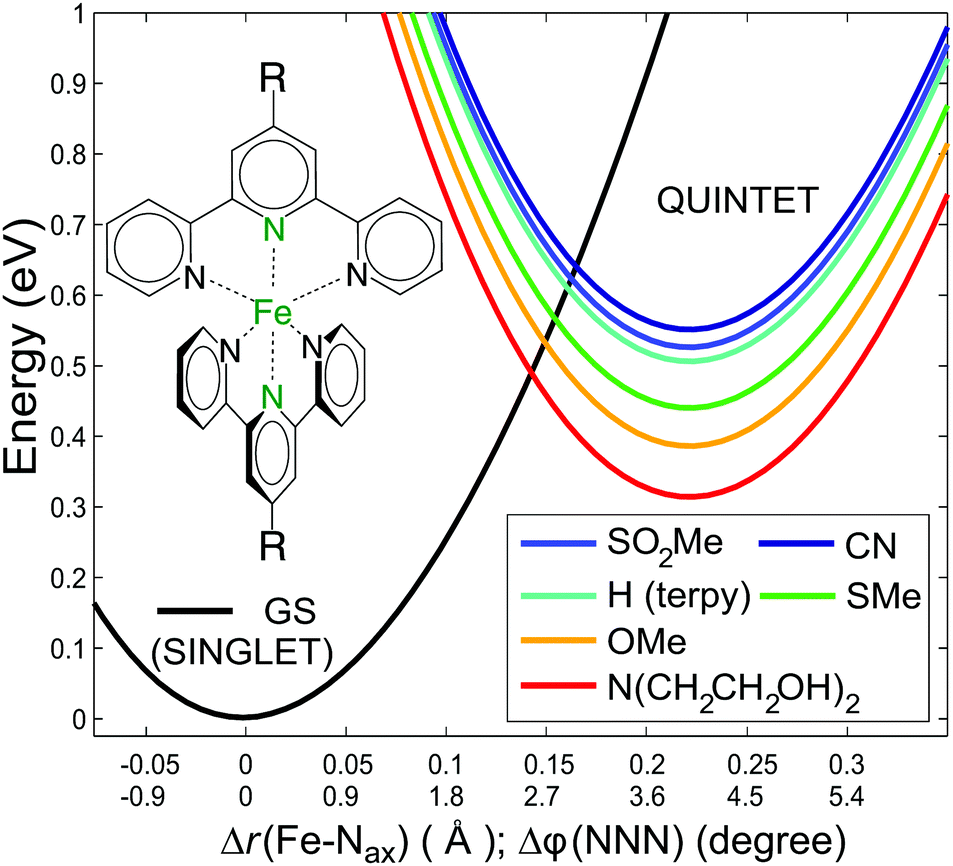 | ||
| Fig. 1 Model potential energy curves for the studied [Fe(terpy)2]2+ derivatives with the quintet energies shifted to the calculated ΔEHL values, plotted along a combined coordinate,10 which connects the singlet and quintet minima. The black curve shows the singlet ground state, while the coloured curves represent the quintet state of the [Fe(4′-R-terpy)2]2+ complex with substituents as indicated in the legend. | ||
While the behavior of complexes with mono- or bidentate ligands is most often described as depending almost exclusively on the (symmetric combination of the) metal–ligand bond lengths, tridentate ligands offer more parameters to control, therefore they seem more promising substances for ligand engineering. Experiments dedicated to elevating the working temperature of such complexes also support this.4 Among them, [Fe(terpy)2]2+ is outstanding because its quintet state is unexpectedly stable under certain circumstances,11,12 which made this complex the subject of intensive studies,10,13,14 together with its derivatives and structural analogs.15–17 Efforts were also made to tune the ligand field in such complexes,16,18 resulting in modifications that led to the first promising low-spin and high-spin Fe(II) chromophores15,17,19 by stabilization of the 3MLCT and 5MLCT states, respectively. (MLCT: metal-to-ligand charge transfer.) Our aim is to exploit new opportunities offered by the complexity of the system to control the quintet lifetime.11,12
The Fe-ligand bond length is one of the most important coordinates in the description of the spin-state switching.3,12 In [Fe(terpy)2]2+, because of geometrical constraints in the ligand, the nitrogen of the central pyridine ring (Nax) is significantly closer to the iron than those of the peripheral rings, that adopt an almost equatorial position in the complex (Neq).13,20 Therefore modifying the electronic structure of the central rings should have a high impact on the bonding along the main axis, significantly reshaping the potential energy surfaces along the highly critical Fe–Nax coordinate.
We have carried out DFT calculations to determine how the ED/EW substitution modifies the energetics related to the quintet–singlet transition in [Fe(4′-R-terpy)2]2+. These calculations reveal that the substitution indeed shifts the energy gap between the singlet and quintet potential energy curves, hence also the barrier for the quintet-singlet relaxation, suggesting increased quintet lifetimes. We tested several complexes of the terpy ligand substituted in the 4′ position, and made a prediction for the variation of the lifetime upon substitution. Measuring the quintet lifetime with transient optical absorption spectroscopy (TOAS) revealed that both shortening and lengthening of the lifetime can be achieved with substitution. Moreover, the observed lifetimes follow well the predicted variations.
DFT has been rather successful in describing the molecular properties of Fe(II) complexes,10,12,21–24 and the B3LYP* functional25 has been found to satisfactorily predict the energetics of spin state transitions.10,21,24,26 We have carried out calculations with the BP8627 and B3LYP* functionals, the TZVP basis set28 and the COSMO solvation model29 with water, on the [Fe(4′-R-terpy)2]2+ complex with R ranging from the EW SO2Me, CN groups to the ED SMe, OMe, N(CH2CH2OH)2. These calculations, performed for the lowest singlet and quintet states using ORCA 3.0,30 are described in detail in the ESI.† For both spin states, the FeN6 core is almost identical for all optimized geometries, and shows no significant correlation with the nature of the substituent group. This is similar to previous findings for the 4-substitution of the pyridine (Py) ring in FeIIPytacn complexes, where ED or EW substituents did not produce relevant variation of Fe–NPy bond lengths.31 On the other hand, the energy difference between the singlet and quintet minima, ΔEHL, shows very substantial variations (Table 1). Strong EW groups, such as SO2Me or CN destabilize the quintet state by 20–50 meV, while ED substituents can cause more substantial stabilization. (The reason behind this is that in the quintet state the molecular orbital based on the 3 dz2 is populated, and this is most affected by the axial substitution.) Such displacement of the quintett minimum means that the potential energy landscape is significantly modified by substitution, and the EW/ED properties can be used to tune the ΔEHL energy gap. It has long been known that this energy gap affects the quintet to singlet relaxation rate: lowering the potential minimum of the quintet leads to a wider barrier between the potential wells, which diminishes tunneling rates at low temperatures, and this has been known as the “inverse energy gap law”.32 At room temperature the relevant parameter is the classical barrier between the quintet and singlet (ΔE‡), which will increase with the decrease in vertical displacement of the potential wells (ΔEHL). The effect on the quintet lifetime can be obtained from the Arrhenius or Eyring-Polanyi equations. The relation between the energy (or Gibbs free energy) barrier and the rate is exponential for both. The lifetime has a reciprocal relation to the relaxation rate, thus it is proportional to exp(ΔE‡/kT).
| R (Subst.) | ΔEHL | ΔE‡X | ΔE‡MECP | τ relX | τ relMECP | τ | τ relexp |
|---|---|---|---|---|---|---|---|
| CN | 551 | 89 | 95 | 0.55 | 0.41 | 1.45(2) | 0.54(2) |
| SO2Me | 525 | 97 | 109 | 0.77 | 0.73 | 1.33(2) | 0.49(1) |
| H | 505 | 103 | 117 | 1 | 1 | 2.69(6) | 1 |
| SMe | 439 | 126 | 153 | 2.5 | 4.2 | 5.92(2) | 2.20(5) |
| OMe | 385 | 146 | 168 | 5.6 | 7.7 | 10.7(1) | 3.98(9) |
| N(C2H5O)2 | 324 | 171 | 191 | 14.8 | 19.3 | 20.8(5) | 7.7(3) |
The effect of the substitution on the quintet lifetime can be estimated by investigating the energy barriers between the quintet and singlet states. The potential energy surfaces for these states of [Fe(terpy)2]2+ were previously calculated with high accuracy at the CASPT2 level over two dimensions spanned by the two most relevant modes, r(Fe–Nax) (the bond length between the Fe and the axial N of the ligand), and the NeqNaxNeq′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :φ(NNN) angle of the ligand.10 The substituted derivatives are expected to exhibit similar potential surfaces, although the energy differences should differ. Using the cross sections of the CASPT2 2D potential energy surfaces for the singlet and quintet states over a reaction coordinate that combines the two modes,10 we plotted in Fig. 1 these potential energy curves, adjusting the ΔEHL from the present calculations for the derivatives studied. It is apparent that the energy barrier between these states is inversely related to the ΔEHL. This qualitative picture allows an educated guess about the relations of the lifetimes if we assume that the Arrhenius equation can be used to approximate the room temperature decay of the quintet state. Barrier heights (ΔE‡i) ranging from 89 meV to 171 meV can be read from the plots in Fig. 1. This barrier was determined from temperature-dependent ultrafast spectroscopy for [Fe(terpy)2]2+ as 94(±9) meV,33 which compares favourably to the 103 meV obtained here. Assuming that the pre-exponential factors do not significantly differ, the relative lifetimes with respect to a selected reference can be expressed with the ratio
:φ(NNN) angle of the ligand.10 The substituted derivatives are expected to exhibit similar potential surfaces, although the energy differences should differ. Using the cross sections of the CASPT2 2D potential energy surfaces for the singlet and quintet states over a reaction coordinate that combines the two modes,10 we plotted in Fig. 1 these potential energy curves, adjusting the ΔEHL from the present calculations for the derivatives studied. It is apparent that the energy barrier between these states is inversely related to the ΔEHL. This qualitative picture allows an educated guess about the relations of the lifetimes if we assume that the Arrhenius equation can be used to approximate the room temperature decay of the quintet state. Barrier heights (ΔE‡i) ranging from 89 meV to 171 meV can be read from the plots in Fig. 1. This barrier was determined from temperature-dependent ultrafast spectroscopy for [Fe(terpy)2]2+ as 94(±9) meV,33 which compares favourably to the 103 meV obtained here. Assuming that the pre-exponential factors do not significantly differ, the relative lifetimes with respect to a selected reference can be expressed with the ratio 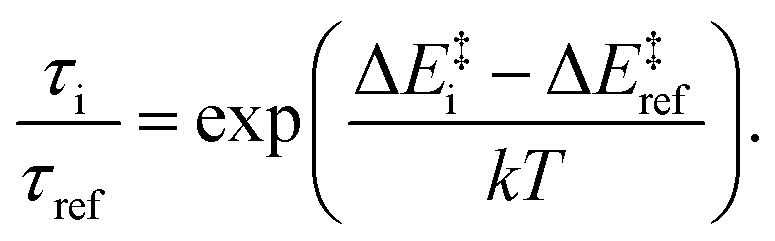
Table 1 displays the above values for all derivatives, where “X” denotes that these values were obtained from the crossing points of the singlet and quintet potential energy curves.
Alternatively the barrier heights can be estimated by locating the minimum energy crossing point (MECP) between the potential energy surfaces of the singlet and quintet states, by performing a geometry optimization minimizing the energy difference between these states.34 The energy between the MECP and the HS minimum corresponds to the barrier (ΔE‡MECP,i). This approach relies solely on calculations on the studied molecule, and takes all possible nuclear degrees of freedom into account. We calculated such barriers at the same level of theory (B3LYP*/TZVP), and also list them in Table 1. The barriers calculated from MECP show a similar relation to ΔEHL: the higher the gap, the smaller the barrier. Moreover the values and their variations for the two types of barriers are in reasonably good agreement. The predicted relative lifetimes computed using the above approach are also listed in the table. The relative lifetimes differ from those of the previous series by a factor of 1.7 in the least favourable case.
In order to test the above concept experimentally, we have prepared the Fe(II) complexes with Cl− counterions and determined the quintet lifetimes. All terpyridine derivatives were synthesized at EvoBlocks Ltd, following literature methods.5,6,35 The preparation and the characterisation of the complexes are described in the ESI.† Purity and composition was verified with LC-HRMS, and 1H NMR. The substituent effects were clearly observed as systematic shifts in the most affected regions of the NMR and UV-VIS spectra, the latter in agreement with time-dependent DFT36 calculations and former experimental observations31 (see the ESI†).
The details of the transient optical absorption measurements are described in the ESI.† In these complexes the relaxation to the long-lived quintet state is completed in a few hundred femtoseconds,12 and the quintet state is essentially optically silent in the window of our white light continuum, so the only signal present after a few ps is the one from the ground state bleach (GSB). This is evident from Fig. 2, which displays the TOAS of the complex with the R = SO2Me substituent (cf. Fig. S3A and S5 in the ESI†).
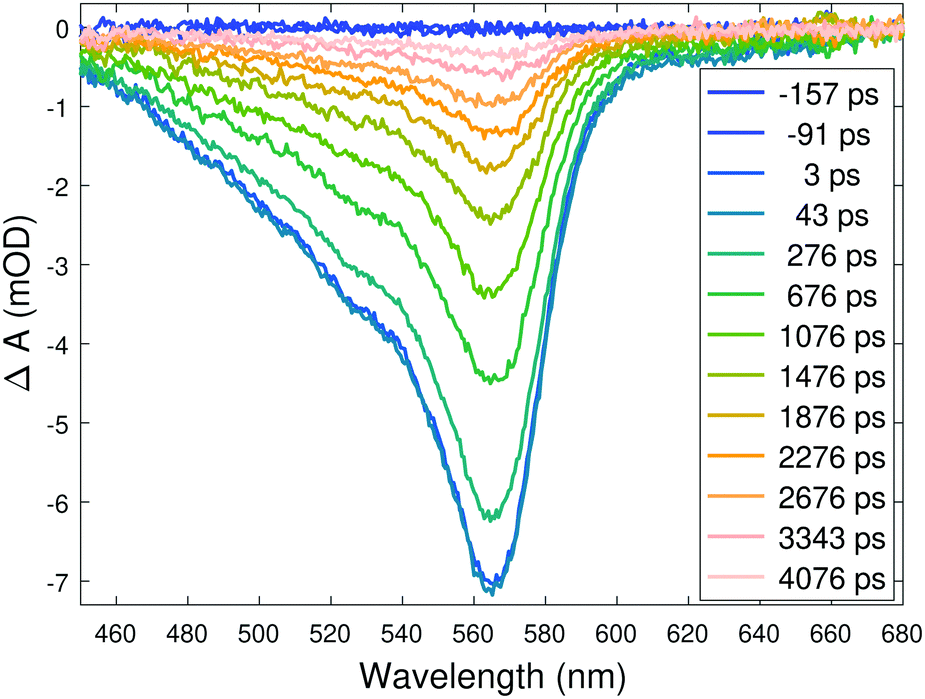 | ||
| Fig. 2 Transient optical absorption spectra of the aqueous solution of the complex with R = SO2Me at selected delays. | ||
As the instrument response function width is below 1 ps, the quintet lifetime can be precisely determined from the GSB recovery, fitting it with an exponential decay, as shown in Fig. 3. The quintet lifetime determined for [Fe(terpy)2]2+ agrees well with previous values in the literature.13,37 The data (Fig. 3) and the fitted lifetimes (Table 1) clearly show the trend that we present as the basis of our main argument.
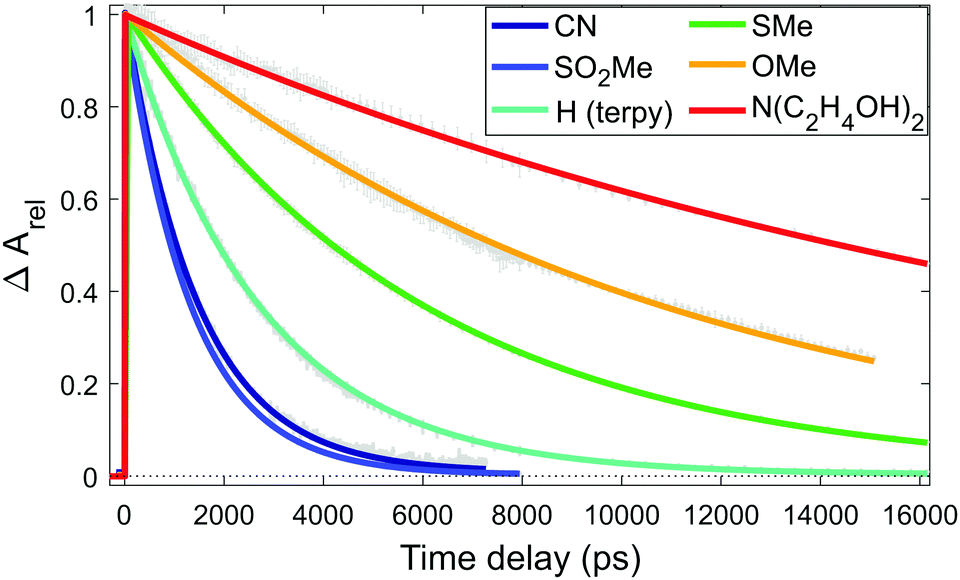 | ||
| Fig. 3 Evolution of the normalized GSB signal for each sample, and their fits with an exponential decay to determine the lifetime. | ||
Comparison of the observed relative lifetimes in Table 1 reveals their agreement with the calculations. The ordering of the two shortest lifetimes (for R = CN and R = SO2Me), appear reversed, otherwise the trend follows the predicted one. The satisfactory correspondence between the a priori theoretical estimates and the observed lifetimes indicates that our approach is able to grasp the main effect of the substitution. We are certainly pushing the limits of the accuracy of DFT, but we can build on the vast literature and our own experience on Fe(II) complexes with the B3LYP* functional that has been particularly successful in describing the energies of the different states. Moreover, we greatly benefit from the high structural and mechanistic similarities of the complexes studied, which allowed us to achieve such accuracy. The good qualitative agreement between the lifetimes predicted by estimating the energy barrier from the shifted potential curves and from locating the minimum energy crossing points on the one hand, and the rather satisfactory agreement of the relative lifetimes predicted by these models with those from our transient optical absorption measurements on the other, should certainly lend credibility to our approach. Our data show that upon substitution of the terpy ligand in the 4′ position of the central ring with EW moieties, the lifetime of the quintet state of its Fe(II) complex is shortened due to destabilization. More important from a practical point of view is that by substitution of the terpy ligand in the same position with ED substituents, a significant, almost 10-fold increase of the quintet lifetime can be achieved.
Innovative ligand design will remain crucial for developing novel functional coordination complexes,6,8,38 and theoretical methods will be indispensable in these efforts. Wave-function-based multireference techniques are expected to deliver precise tools for this, but theory can already provide invaluable insight with acceptable accuracy at affordable cost at the DFT level.23,39 We successfully demonstrated that quantum-chemistry-based ligand engineering of Fe complexes in particular, and of transition-metal-based functional molecules in general, can have great potential in attaining the desired properties and behavior, which can be exploited in the future in a plethora of applications.
This project was supported by the “Lendület” (Momentum) Program of the Hungarian Academy of Sciences (LP2013-59), the Government of Hungary and the European Regional Development Fund under grant VEKOP-2.3.2-16-2017-00015. We thank Dr G. Bazsó for help in realizing the instrument control, Dr P. Rácz for assistance with the laser, Dr L. Burai and Dr G. Orgován (Servier Hungária) for the LC-HRMS measurements, A. Vancza for recording the UV-VIS spectra, Dr A. Domján for recording the NMR spectra, and Dr T. Rozgonyi for his comments on the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) O. Kahn and C. J. Martinez, Science, 1998, 279, 44 CrossRef; (b) G. Benkö, et al. , J. Am. Chem. Soc., 2002, 124, 489 CrossRef; (c) A. Bousseksou, et al. , Chem. Soc. Rev., 2011, 40, 3313 RSC; (d) C. A. P. Goodwin, et al. , Nature, 2017, 548, 439 CrossRef; (e) K. S. Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176 CrossRef.
- S. Decurtins, et al. , Chem. Phys. Lett., 1984, 105, 1 CrossRef.
- A. Hauser, J. Chem. Phys., 1991, 94, 2741 CrossRef.
- J.-F. Létard, et al. , Chem. – Eur. J., 2005, 11, 4582 CrossRef.
- (a) E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133 RSC; (b) J. Guérin, et al. , J. Lumin., 2011, 131, 2668 CrossRef; (c) K. Nakano, et al. , Dalton Trans., 2005, 740 RSC.
- A. K. Pal and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 6184 RSC.
- (a) B. J. Houghton and R. J. Deeth, Eur. J. Inorg. Chem., 2014, 4573 CrossRef; (b) D. C. Ashley and E. Jakubikova, Inorg. Chem., 2018, 57, 9907 CrossRef.
- M. A. Halcrow, Crystals, 2016, 6, 58 CrossRef.
- L. J. Kershaw Cook, et al. , Angew. Chem., Int. Ed., 2016, 55, 4327 CrossRef CAS.
- M. Pápai, et al. , J. Chem. Theory Comput., 2013, 9, 509 CrossRef.
- F. Renz, et al. , Angew. Chem., Int. Ed., 2000, 39, 3699 CrossRef.
- A. Hauser, et al. , Coord. Chem. Rev., 2006, 250, 1642 CrossRef.
- G. Vankó, et al. , J. Phys. Chem. C, 2015, 119, 5888 CrossRef.
- (a) S. E. Canton, et al. , J. Phys. Chem. C, 2014, 118, 4536 CrossRef; (b) X. Zhang, et al. , J. Phys. Chem. C, 2015, 119, 3312 CrossRef CAS; (c) J. Nance, et al. , Inorg. Chem., 2015, 54, 11259 CrossRef CAS; (d) S. E. Canton, et al. , J. Phys. Chem. C, 2015, 119, 3322 CrossRef CAS; (e) A. M. March, et al. , J. Phys. Chem. C, 2017, 121, 2620 CrossRef CAS.
- Y. Liu, et al. , Chem. Commun., 2013, 49, 6412 RSC.
- L. L. Jamula, et al. , Inorg. Chem., 2014, 53, 15 CrossRef CAS.
- (a) S. G. Shepard, et al. , J. Am. Chem. Soc., 2016, 138, 2949 CrossRef CAS; (b) S. M. Fatur, et al. , J. Am. Chem. Soc., 2017, 139, 4493 CrossRef CAS; (c) Z. S. Yoon, et al. , J. Phys. Chem. B, 2010, 114, 11731 CrossRef CAS; (d) L. J. Kershaw Cook, et al. , Dalton Trans., 2013, 42, 2254 RSC; (e) I. M. Dixon, et al. , Inorg. Chem., 2013, 52, 13369 CrossRef CAS; (f) C. R. Benson, et al. , Dalton Trans., 2014, 43, 6513 RSC; (g) I. M. Dixon, et al. , Dalton Trans., 2015, 44, 13498 RSC; (h) M. Wächtler, et al. , Phys. Chem. Chem. Phys., 2016, 18, 2350 RSC.
- (a) E. C. Constable, et al. , Inorg. Chim. Acta, 1998, 275-276, 359 CrossRef; (b) E. C. Constable, et al. , Chem. – Eur. J., 1999, 5, 498 CrossRef; (c) D. N. Bowman, et al. , Inorg. Chem., 2015, 54, 8786 CrossRef.
- (a) T. C. B. Harlang, et al. , Nat. Chem., 2015, 7, 883 CrossRef; (b) M. Pápai, et al. , J. Phys. Chem. Lett., 2016, 7, 2009 CrossRef.
- A. T. Baker and H. A. Goodwin, Aust. J. Chem., 1985, 38, 207 CrossRef.
- M. Reiher, Inorg. Chem., 2002, 41, 6928 CrossRef.
- H. Paulsen and A. X. Trautwein, Top. Curr. Chem., 2004, 235, 197 CrossRef.
- D. C. Ashley and E. Jakubikova, Coord. Chem. Rev., 2017, 337, 97 CrossRef.
- A. Vargas, et al. , J. Chem. Theory Comput., 2009, 5, 97 CrossRef.
- (a) M. Reiher, Theor. Chem. Acc., 2001, 107, 48 Search PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) C. Lee, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (d) P. J. Stephens, et al. , J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- (a) J. N. Harvey, in DFT Computation of Relative Spin-State Energetics of Transition Metal Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 151 Search PubMed; (b) L. M. Lawson Daku, et al. , ChemPhysChem, 2005, 6, 1393 CrossRef; (c) Y. Shiota, et al. , J. Phys. Chem. A, 2010, 114, 5862 CrossRef; (d) K. P. Kepp, Inorg. Chem., 2016, 55, 2717 CrossRef; (e) V. I. Minkin, et al. , Dalton Trans., 2016, 45, 12103 RSC; (f) D. Leshchev, et al. , Chem. Sci., 2018, 9, 405 RSC.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- (a) A. Schäfer, et al. , J. Chem. Phys., 1992, 97, 2571 CrossRef; (b) A. Schäfer, et al. , J. Chem. Phys., 1994, 100, 5829 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- I. Prat, et al. , Inorg. Chem., 2013, 52, 9229 CrossRef CAS.
- (a) A. Hauser, et al. , J. Chem. Phys., 1991, 95, 8710 CrossRef CAS; (b) P. Gütlich, et al. , Angew. Chem., Int. Ed. Engl., 1994, 33, 2024 CrossRef.
- M. C. Carey, et al. , Chem. Sci., 2019, 10, 134 RSC.
- (a) J. N. Harvey, et al. , Theor. Chem. Acc., 1998, 99, 95 Search PubMed; (b) T. Chachiyo and J. H. Rodriguez, J. Chem. Phys., 2005, 123, 094711 CrossRef; (c) J. N. Harvey, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 1 CAS.
- (a) D. L. Jameson and L. E. Guise, Tetrahedron Lett., 1991, 32, 1999 CrossRef CAS; (b) K. T. Potts, et al. , J. Am. Chem. Soc., 1987, 109, 3961 CrossRef CAS; (c) J. Husson, et al. , Nat. Protoc., 2014, 9, 21 CrossRef CAS; (d) U. S. Schubert, et al., Modern Terpyridine Chemistry, Wiley-VCH, 2006 CrossRef.
- (a) F. Neese and G. Olbrich, Chem. Phys. Lett., 2002, 362, 170 CrossRef CAS; (b) T. Petrenko, et al. , J. Chem. Phys., 2011, 134, 054116 CrossRef.
- C. Creutz, et al. , J. Am. Chem. Soc., 1980, 102, 1309 CrossRef CAS.
- (a) D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC; (b) P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591 CrossRef CAS.
- F. Neese, Angew. Chem., Int. Ed., 2017, 56, 11003 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational and experimental details, UV-vis, NMR spectra, LC-HRMS data, optimized geometries. See DOI: 10.1039/d0cc04467a |
| This journal is © The Royal Society of Chemistry 2020 |