
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and hetero-Diels–Alder reactions of enantiomerically pure dihydro-1H-azepines†
Donald
Craig
 *a,
Samuel R. J.
Spreadbury
a and
Andrew J. P.
White
b
*a,
Samuel R. J.
Spreadbury
a and
Andrew J. P.
White
b
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: d.craig@imperial.ac.uk; Tel: +44 (0)20 7594 5771
bChemical Crystallography Laboratory, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK
First published on 14th July 2020
Abstract
Thermolysis of enantiomerically pure 3-substituted 7,7-dihalo-2-azabicyclo[4.1.0]heptanes in the presence of K2CO3 gives in good yields 2-alkyl-6-halo-1-tosyl-2,3-dihydro-1H-azepines. These undergo highly stereoselective [4+2] cycloaddition reactions with heterodienophiles and arylation/alkenylation under Suzuki conditions.
Stereodefined azepine and azepane derivatives are valuable molecular scaffolds present in several bioactive natural products and pharmaceutically relevant molecules.1 Species featuring these seven-membered heterocyclic cores and related compounds have received considerable attention because of their potential as glycosidase inhibitors2 and anticancer,3 anti-diabetic4 and anti-viral agents.5 Consequently, a number of methodologies have been developed for their preparation, with recent accounts detailing ring-expansion cascades,6 cycloaddition approaches7 and cyclisation strategies.8
The importance of gem-dihalocyclopropanes in synthesis stems in large part from their ready accessibility and high reactivity in a range of transformations.9 More specifically, a number of synthesis methodologies that have been developed exploit the reactivity of cyclopropanes bearing nitrogen substituents, most notably involving ring-opening and rearrangement processes.10 Of particular interest to us were the largely unexplored thermal ring-expansion reactions of gem-dihalocyclopropanes in which the three-membered ring was fused to an N-heterocycle. These often low-yielding processes required high temperatures or activation by silver(I) salts to generate the putative allylic cationic intermediates, which were typically intercepted by alcohols or hydride reagents to afford vinyl halides.11 To date, there have been few reports of the successful isolation of diene products in the absence of a nucleophilic additive or solvent.12
Our laboratory has previously investigated the synthesis and chemistry of N-arylsulfonyl-1,2,3,4-tetrahydropyridines, in particular the utility of these for the stereoselective elaboration of more complex N-heterocycles.13 It occurred to us that bicyclo[4.1.0] products of dihalocyclopropanation of enantiomerically pure N-arylsulfonyl-1,2,3,4-tetrahydropyridines would undergo ring-opening and deprotonation to give stereodefined dihydroazepines. In this work, we describe base-mediated thermal ring-expansion reactions of 3-substituted 7,7-dihalo-2-azabicyclo[4.1.0]heptanes to give halogenated dihydro-1H-azepines, and present stereoselective [4+2] cycloaddition and Pd(0)-catalysed cross-coupling reactions of these novel scaffolds.
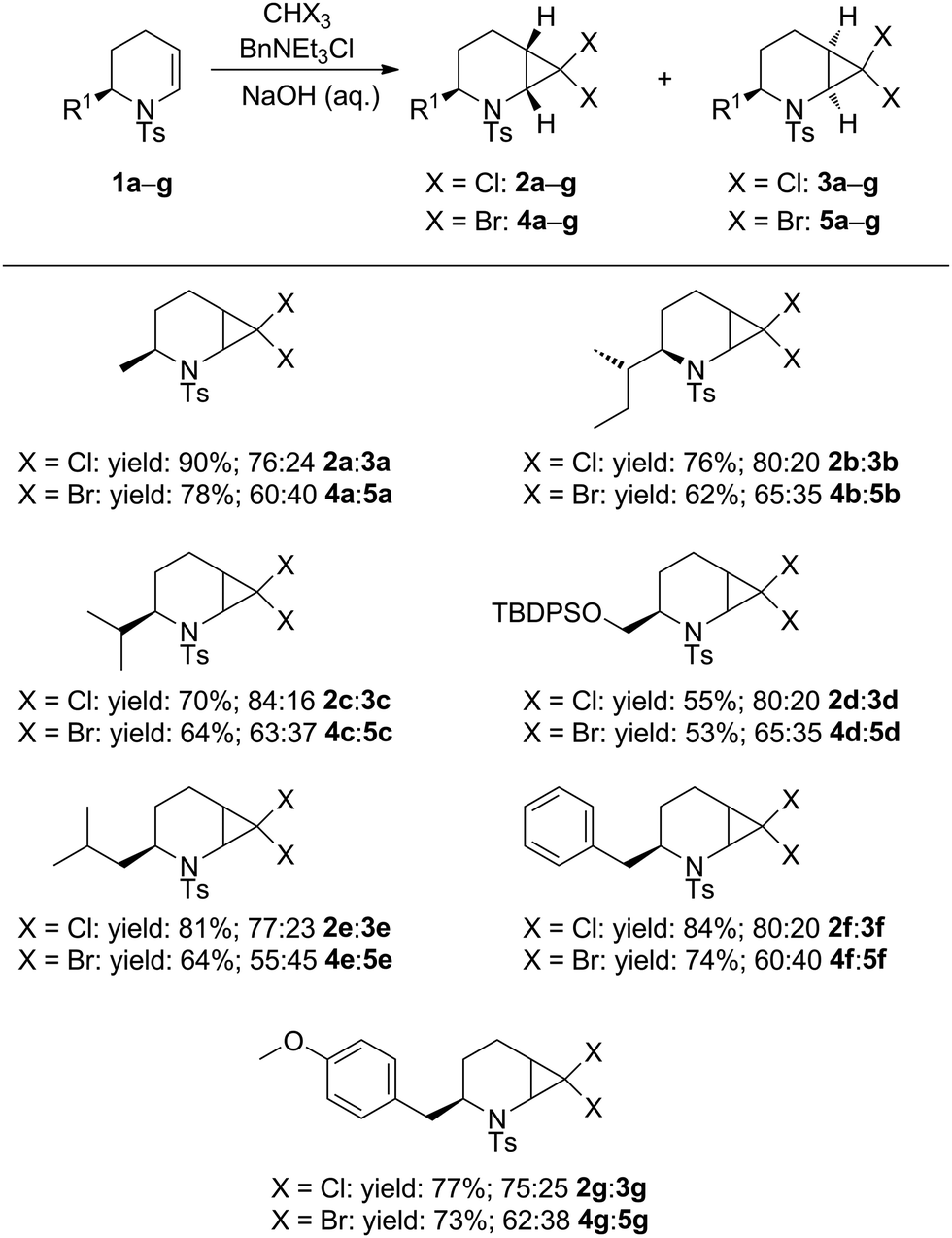
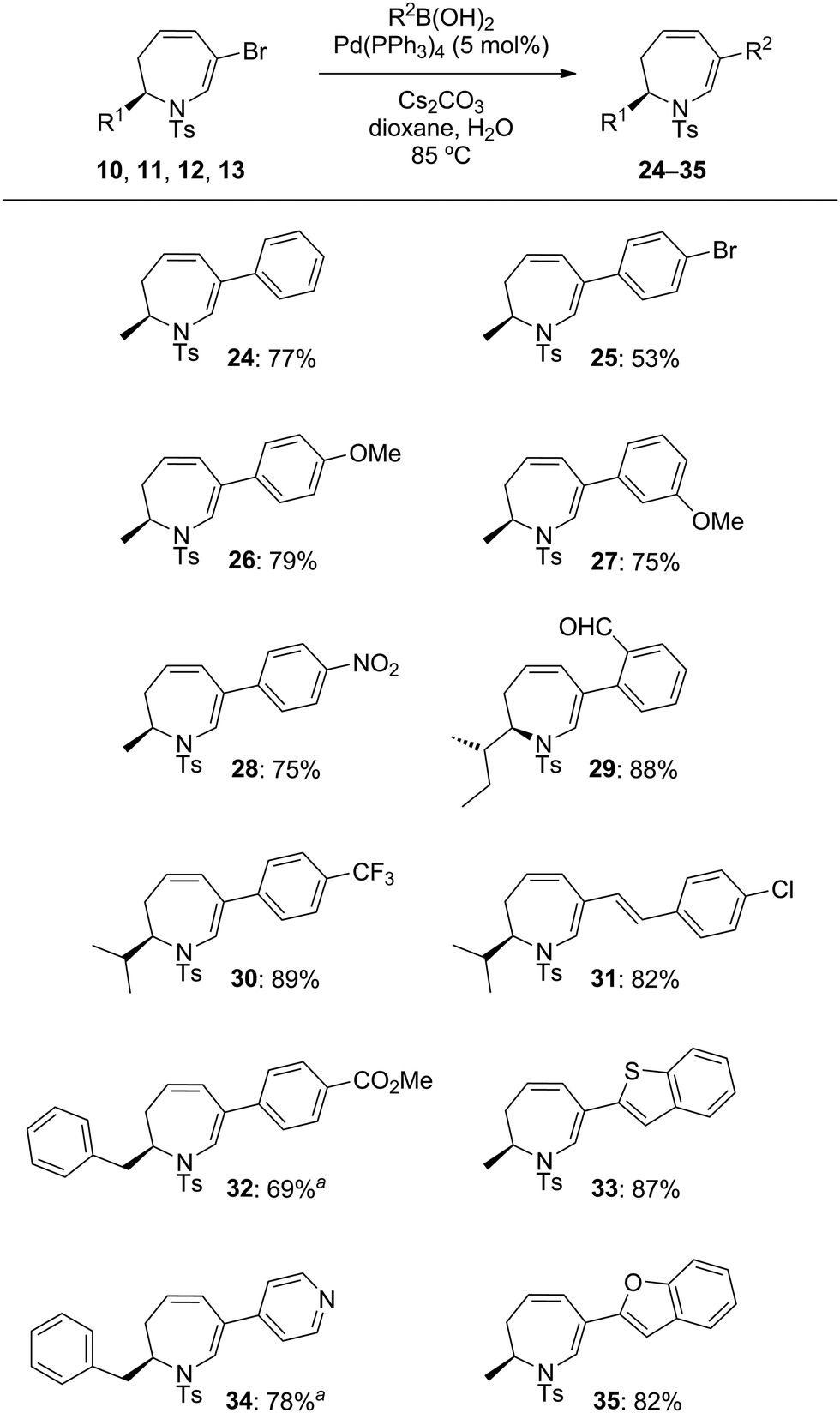
The enantiomerically pure 2-substituted 1,2,3,4-tetrahydropyridines 1a–g used in this study were synthesised according to the procedure of Harrity and co-workers14 from L-amino acid-derived N-tosylaziridines.15 Substrates 1a–g were subjected to dihalocyclopropanation under conditions reported by Mąkosza,16 giving dichloro- and dibromo-substituted 2-azabicyclo[4.1.0]heptanes 2a–g, 3a–g and 4a–g, 5a–g, respectively as anti/syn mixtures in good to excellent yields. For examples a (R1 = Me), d (R1 = TBDPSOCH2), f (R1 = Bn), and g (R1 = 4-MeOC6H4CH2), compounds 2/3 (X = Cl) and 4/5 (X = Br) were obtained as inseparable anti/syn mixtures (Table 1).
|
Moderate stereoselectivity for the anti diastereoisomers 2 and 4 was observed for all the substrates studied, as was indicated by 1H NMR analysis and assigned unambiguously by X-ray crystallographic analysis of 2a and 3a. Similar facial selectivity in the dichlorocyclopropanation of cyclic enamides has been reported recently.17 We postulate that the lower anti-selectivity observed in the dibromocarbene addition reactions is a consequence of the greater steric interaction of the carbene with the N-tosyl group, which adopts a conformation anti to the R substituent.13
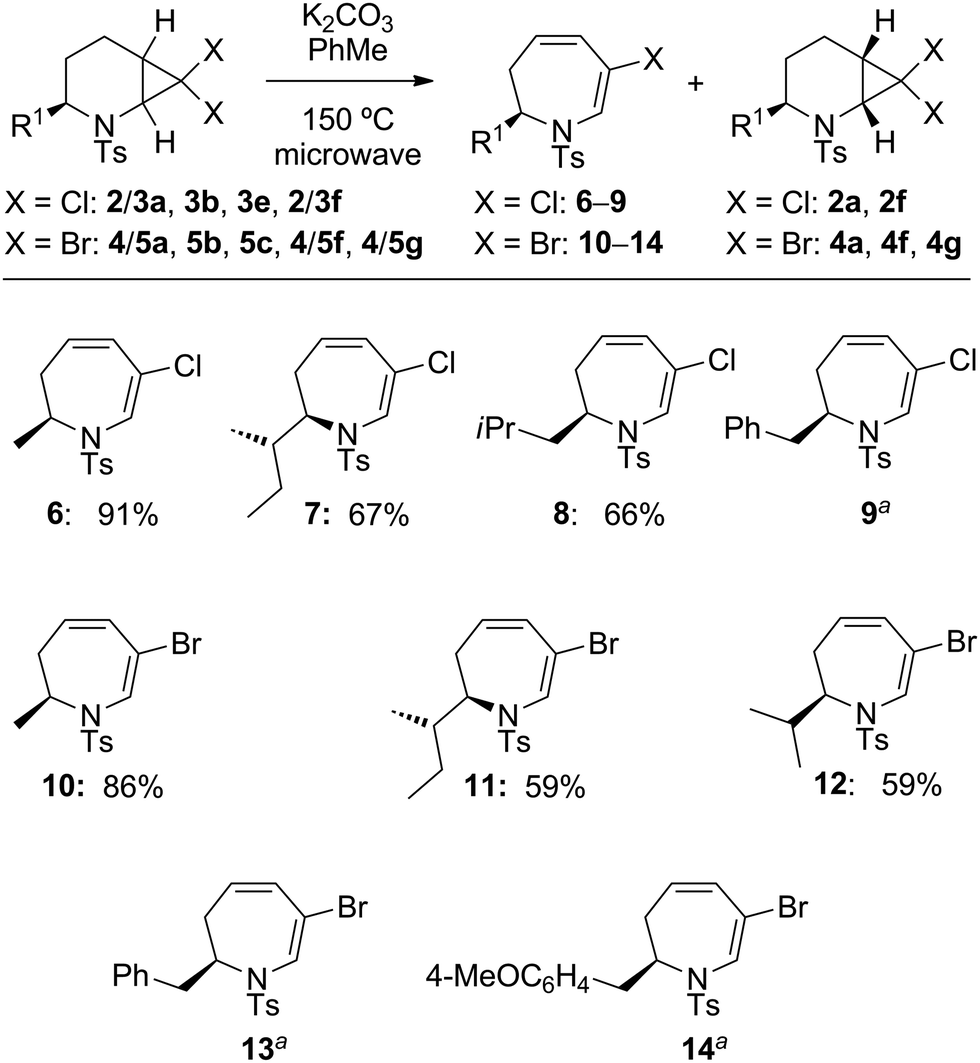
Initial ring-expansion experiments involved exposure of the ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2a + 3a to varying combinations of base and silver salts (see ESI† for optimisation conditions). Although no consumption of substrate was observed at ambient temperature, the use of microwave irradiation at 150 °C resulted in conversion of only the syn diastereoisomer 3a into the desired dihydro-1H-azepine 6. Further investigation revealed that Ag(I) additives were unnecessary and that the addition of one equivalent of potassium carbonate in toluene at 150 °C for 5 hours under microwave irradiation conditions resulted in improved yields of 6, although the anti diastereoisomer 2a still failed to react under these modified conditions. Several additional 7,7-dihalo-2-azabicyclo[4.1.0]heptanes were subjected to the optimised ring-expansion reaction conditions either as pure syn isomers 3 (3b, 3e: R1 = s-Bu, i-Bu) and 5 (5b, 5e: R1 = s-Bu, i-Pr) or as anti/syn mixtures of 2/3 (2/3a, 2/3f: R1 = Me, Bn) and 4/5 (4/5a, 4/5f, 4/5g: R1 = Me, Bn, 4-MeOC6H4CH2) to give dihydroazepines 6–14 in good to excellent yields based on the syn isomers 3 and 5 (Table 2). Dihydroazepines 9, 13 and 14 were inseparable from the unreacted anti substrates 2f, 4f and 4g, respectively.
:1 mixture of 2a + 3a to varying combinations of base and silver salts (see ESI† for optimisation conditions). Although no consumption of substrate was observed at ambient temperature, the use of microwave irradiation at 150 °C resulted in conversion of only the syn diastereoisomer 3a into the desired dihydro-1H-azepine 6. Further investigation revealed that Ag(I) additives were unnecessary and that the addition of one equivalent of potassium carbonate in toluene at 150 °C for 5 hours under microwave irradiation conditions resulted in improved yields of 6, although the anti diastereoisomer 2a still failed to react under these modified conditions. Several additional 7,7-dihalo-2-azabicyclo[4.1.0]heptanes were subjected to the optimised ring-expansion reaction conditions either as pure syn isomers 3 (3b, 3e: R1 = s-Bu, i-Bu) and 5 (5b, 5e: R1 = s-Bu, i-Pr) or as anti/syn mixtures of 2/3 (2/3a, 2/3f: R1 = Me, Bn) and 4/5 (4/5a, 4/5f, 4/5g: R1 = Me, Bn, 4-MeOC6H4CH2) to give dihydroazepines 6–14 in good to excellent yields based on the syn isomers 3 and 5 (Table 2). Dihydroazepines 9, 13 and 14 were inseparable from the unreacted anti substrates 2f, 4f and 4g, respectively.
| a Compounds 9, 13, 14 were obtained as inseparable mixtures with unreacted anti substrates 2f, 4f and 4g, respectively. |
|---|
|
The difference in ring-expansion reactivity between the syn isomers 3/5 and the anti isomers 2/4 is striking. Inspection of the obtained crystal structures for 2a and 3a indicates a greater degree of nitrogen pyramidalisation in the syn-isomer 3a than in the anti-isomer 2a (see X-ray ESI†). We speculate that this increases the availability of the nitrogen lone pair in 3 to participate in cyclopropane ring opening (Scheme 1).
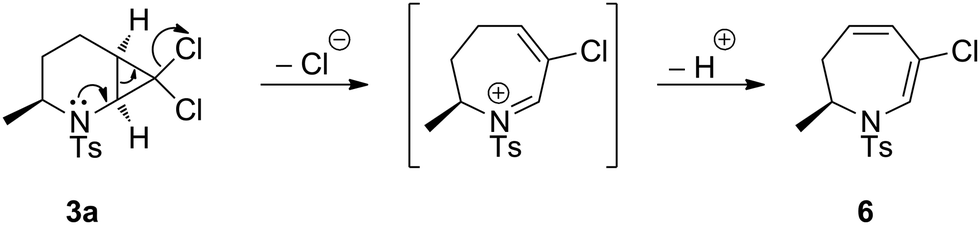 | ||
| Scheme 1 Proposed mechanism for ring-opening of 3a. | ||
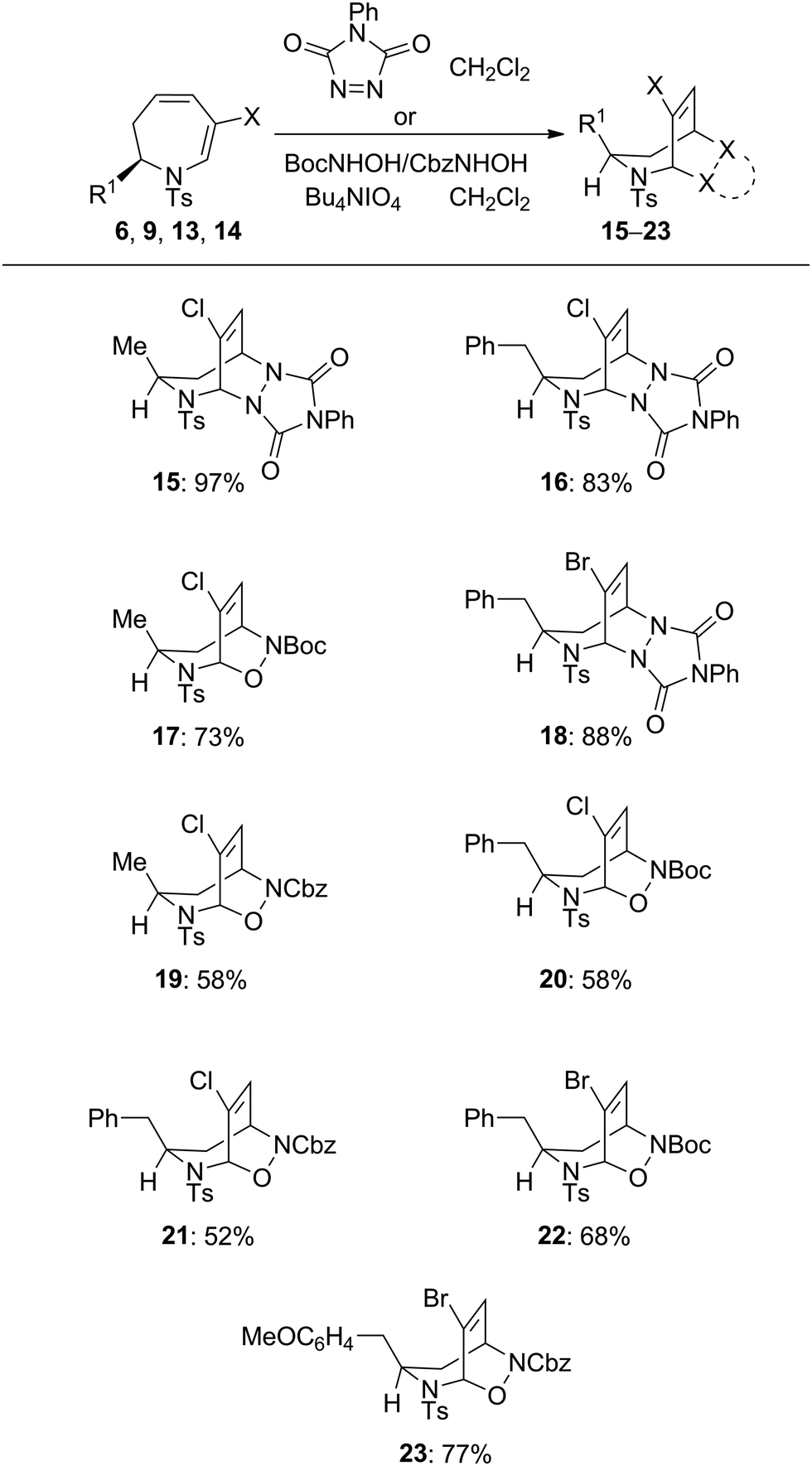
The cycloaddition reactivity of the enantiomerically pure dihydroazepines was investigated next. Combination of analogues 6, 9, 13 and 1418 at ambient temperature with the highly reactive heterodienophiles 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione, and tert-butyl or benzyl nitrosoformate generated in situ by Bu4NIO4-mediated oxidation of the corresponding alkyl hydroxycarbamates gave in excellent yields the products of [4+2] heterocycloaddition, exclusively anti with respect to the R1 substituent on the seven-membered ring (Table 3). The stereochemistry of the cycloadducts 15 and 23 was unequivocally established by X-ray crystallographic analysis (Fig. 1), which demonstrated also the complete regioselectivity of formation of the benzyl nitrosoformate adduct 23.
| a Compounds 9, 13, 14 were used as inseparable mixtures with unreacted 2f, 4f and 4g, respectively; yields of 16, 18, 20–23 are based on the calculated amount of the dihydroazepines in the mixtures (1H NMR). |
|---|
|
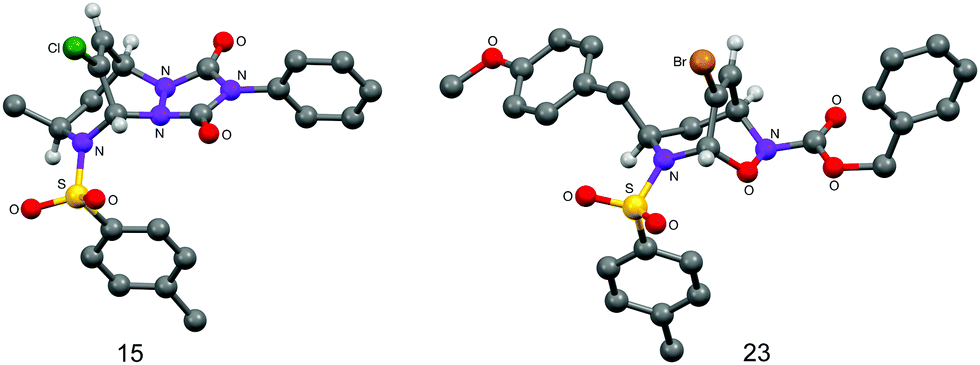 | ||
| Fig. 1 The crystal structures of 15 and 23. | ||
The last part of this study looked at the functionalisation of bromo-substituted 6-bromo-2,3-dihydro-1H-azepines using Pd-catalysed cross-coupling reactions. Substrates 10–13 were coupled with a range of electron-rich and electron-poor arylboronic acids under Suzuki–Miyaura conditions to give the 6-arylated analogues in excellent yields (Table 4).19 On combination with excess 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione in CH2Cl2 at ambient temperature, the triene product 31 entered into hetero-Diels–Alder reaction to give in 87% yield a chromatographically separable mixture of the mono- and bis-adducts 36 and 37 in a 1:2.5 ratio (Scheme 2). The stereochemistry of mono-adduct 36 was inferred from the stereoselectivity observed in the hetero-Diels–Alder reactions of 6, 9, 13 and 14; that of the bis-adduct 37 was established by X-ray crystallographic analysis (Fig. 2).
| a 2-Benzyl-6-bromo-1-tosyl-2,3-dihydro-1H-azepine 13 was used in these reactions as an inseparable mixture with unreacted 4f; yields for products 32 and 34 are for the two steps from the 4f/5f mixture based on the calculated amount of 5f (1H NMR). |
|---|
|
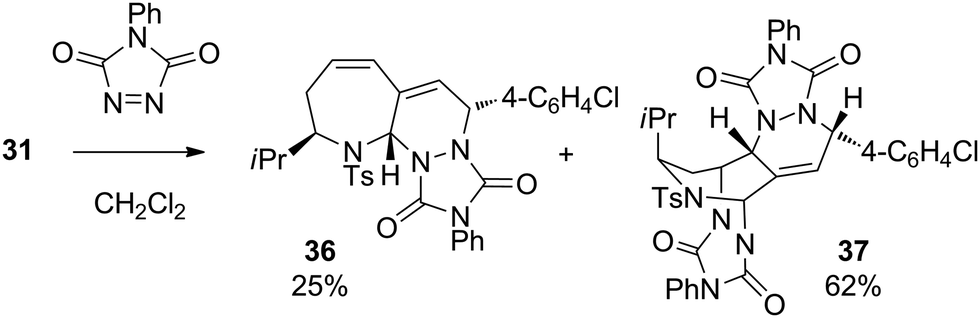 | ||
| Scheme 2 Formation of 36 and 37. | ||
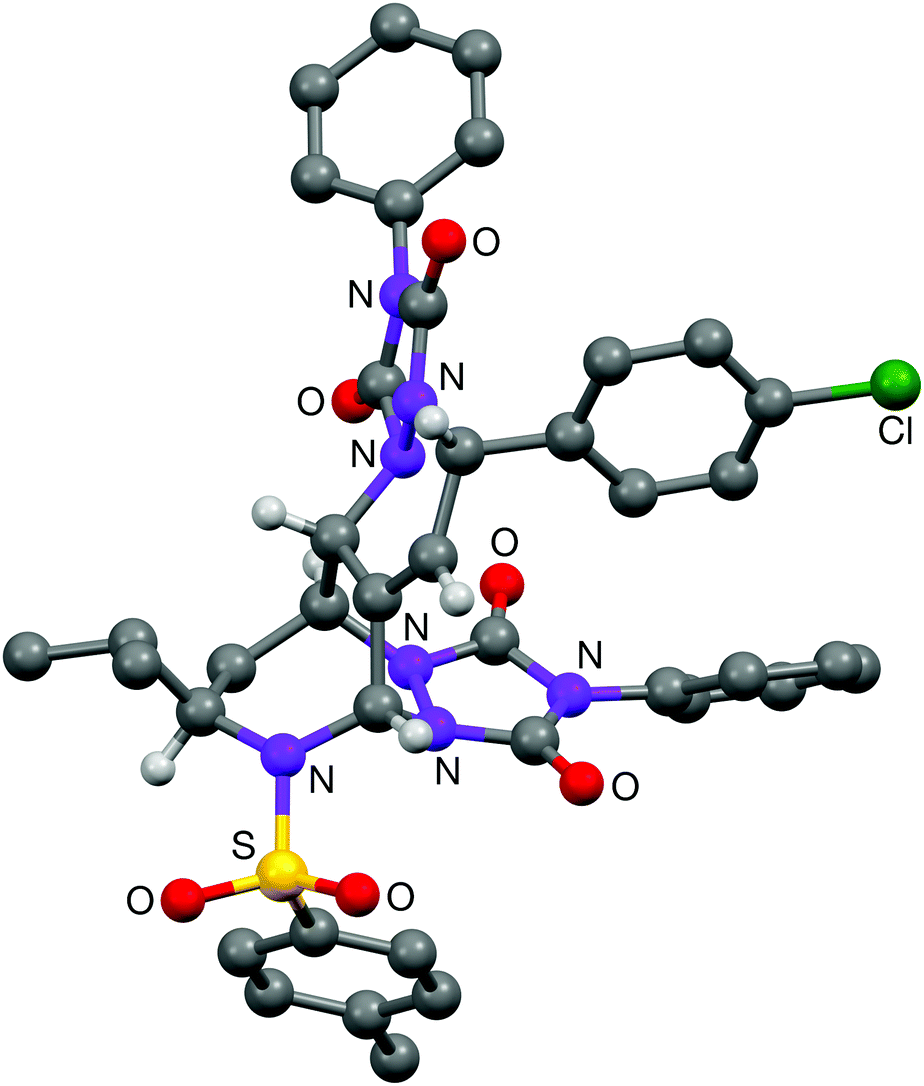 | ||
| Fig. 2 The structure of 37-A, one of the two independent molecules present in the crystal of 37. | ||
This selectivity demonstrated the expected greater intrinsic reactivity of the s-cis cyclic diene in 31 with respect to the conformationally more flexible styryl-containing endocyclic/exocyclic moiety.
In conclusion, we have developed an efficient ring-expansion sequence for the conversion of stereodefined 7,7-dihalo-2-azabicyclo[4.1.0]heptanes into enantiomerically pure 2,3-dihydro-1H-azepines. These molecular scaffolds undergo hetero-Diels–Alder cycloadditions with high stereoselectivity and complete regioselectivity. Additionally, these entities can be efficiently elaborated with a range of aromatic substituents using Suzuki coupling reactions. Further investigation into dihydroazepine derivatisation and application of this chemistry to natural and unnatural product synthesis is ongoing.
We thank EPSRC (Doctoral Training Programme: PhD Studentship to S. R. J. S.) for support.
Conflicts of interest
The authors have no conflicts of interest to declare.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (b) G. F. Zha, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam and S. Long, Eur. J. Med. Chem., 2019, 162, 465 CrossRef CAS PubMed.
- (a) F. Moris-Varas, X. H. Qian and C. H. Wong, J. Am. Chem. Soc., 1996, 118, 7647 CrossRef CAS; (b) G. F. Painter, P. J. Eldridge and A. Falshaw, Bioorg. Med. Chem., 2004, 12, 225 CrossRef CAS PubMed.
- (a) B. Winchester and G. W. J. Fleet, Glycobiology, 1992, 2, 199 CrossRef CAS PubMed; (b) Y. Nishimura, Curr. Top. Med. Chem., 2003, 3, 575 CrossRef CAS PubMed.
- (a) H. Li, Y. Zhang, P. Vogel, P. Sinaÿ and Y. Blériot, Chem. Commun., 2007, 183 RSC; (b) B. Luo, F. Marcelo, J. Désire, Y. Zhang, M. Sollogoub, A. Kato, I. Adachi, F. Javier Cañada, J. Jiménez-Barbero and Y. Blériot, J. Carbohyd. Chem., 2011, 30, 641 CrossRef CAS.
- (a) P. Greimel, J. Spreitz, A. E. Stütz and T. M. Wrodnigg, Curr. Top. Med. Chem., 2003, 3, 513 CrossRef CAS PubMed; (b) H. Li, Y. Blériot, C. Chantereau, J. M. Mallet, M. Sollogoub, Y. Zhang, E. Rodríguez-García, P. Vogel, J. Jiménez-Barbero and P. Sinaÿ, Org. Biomol. Chem., 2004, 2, 1492 RSC; (c) I. Robina, A. J. Moreno-Vargas, A. T. Carmona and P. Vogel, Curr. Drug Metab., 2004, 5, 329 CrossRef CAS PubMed.
- (a) J. Zhou and Y. Y. Yeung, Org. Lett., 2014, 16, 2134 CrossRef CAS PubMed; (b) A. Nortcliffe and C. J. Moody, Bioorg. Med. Chem., 2015, 23, 2730 CrossRef CAS PubMed; (c) J. Y. Du, X. T. An, X. H. Zhao, X. Y. Ma, Y. X. Cao and C. A. Fan, Tetrahedron, 2019, 75, 1760 CrossRef CAS.
- (a) C. Hu, R. J. Song, M. Hu, Y. Yang, J. H. Li and S. Luo, Angew. Chem., Int. Ed., 2016, 55, 10423 CrossRef CAS PubMed; (b) C. Z. Zhu, J. J. Feng and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 1351 CrossRef CAS PubMed; (c) V. Motornov and P. Beier, J. Org. Chem., 2018, 83, 15195 CrossRef CAS PubMed; (d) D. Singh and H. J. Ha, Org. Biomol. Chem., 2019, 17, 3093 RSC; (e) X. Li, S. Wang, S. Li, K. Li, X. Mo, L. Liu, W. Chang and J. Li, J. Org. Chem., 2019, 84, 1288 CrossRef CAS PubMed.
- (a) E. Cini, G. Bifulco, G. Menchi, M. Rodriquez and M. Taddei, Eur. J. Org. Chem., 2012, 2133 CrossRef CAS; (b) W. Zhu, L. Zhao and M. X. Wang, J. Org. Chem., 2015, 80, 12047 CrossRef CAS PubMed; (c) A. Barbero, A. Diez-Varga, F. J. Pulido and A. Gonzàlez-Ortega, Org. Lett., 2016, 18, 1972 CrossRef CAS PubMed; (d) G. W. Wang and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 2743 CrossRef CAS PubMed; (e) A. Artigas, J. Vila, A. Lledó, M. Solà, A. Pla-Quintana and A. Roglans, Org. Lett., 2019, 21, 6608 CrossRef CAS PubMed.
- (a) B. Halton and J. Harvey, Synlett, 2006, 1975 CAS; (b) A. P. Thankachan, K. S. Sindhu, K. K. Krishnan and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 8780 RSC.
- V. A. Rassadin and Y. Six, Tetrahedron, 2016, 72, 4701 CrossRef CAS.
- (a) C. D. Perchonock, I. Lanos, J. A. Finkelstein and K. G. Holden, J. Org. Chem., 1980, 45, 1950 CrossRef CAS; (b) H. P. Soetens and U. K. Pandit, Recl. Trav. Chim. Pay-B, 1980, 98, 271 Search PubMed; (c) D. Dhanak, R. Kuroda and C. B. Reese, Tetrahedron Lett., 1987, 28, 1827 CrossRef CAS.
- (a) T. Inoue, S. Yokoshima and T. Fukuyama, Heterocycles, 2009, 79, 373 CrossRef CAS; (b) G. Chen, P. Kattanguru, O. A. Tomashenk, R. Karpowicz, G. Siemiaszko, A. Bhattacharya, V. Calasans and Y. Six, Org. Biomol. Chem., 2017, 15, 5364 RSC.
- (a) D. Craig, R. McCague, G. A. Potter and M. R. V. Williams, Synlett, 1998, 55 CrossRef CAS; (b) D. Craig, R. McCague, G. A. Potter and M. R. V. Williams, Synlett, 1998, 58 CrossRef CAS; (c) J. C. Adelbrecht, D. Craig, B. W. Dymock and S. Thorimbert, Synlett, 2000, 467 CAS; (d) J. C. Adelbrecht, D. Craig, A. J. Fleming and F. M. Martin, Synlett, 2005, 2643 CAS.
- L. C. Pattenden, R. A. J. Wybrow, S. A. Smith and J. P. A. Harrity, Org. Lett., 2006, 8, 3089 CrossRef CAS PubMed.
- M. B. Berry and D. Craig, Synlett, 1992, 41 CrossRef CAS . Standard procedures for the synthesis of 1 from α-amino acids are provided in the ESI†.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) kosza and M. Wawrzyniewicz, Tetrahedron Lett., 1969, 10, 4659 CrossRef.
kosza and M. Wawrzyniewicz, Tetrahedron Lett., 1969, 10, 4659 CrossRef. - T. J. Idzik, Z. M. Myk and J. G. Sośnicki, J. Org. Chem., 2019, 84, 8046 CrossRef CAS PubMed.
- In the hetero-Diels–Alder reactions 2,3-dihydro-1H-azepines 9, 13 and 14 were used as inseparable mixtures with the unreacted anti-7,7-dihalo-2-azabicyclo[4.1.0]heptanes 2f, 4f and 4g, respectively.
- Bromo-substituted hetero-Diels–Alder adducts 18 and 22 also were found to be effective substrates in Suzuki reactions, giving arylated products in 75–98% yield. Full experimental and spectroscopic details are provided in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, spectroscopic and X-ray crystallographic data (for 2b/3b, 15, 23, 37). CCDC 2011985–2011988. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04413j |
| This journal is © The Royal Society of Chemistry 2020 |