
Hydrogenation and electrocatalytic reduction of carbon dioxide to formate with a single Co catalyst†
Fang
Wang
 ,
Austin T.
Cannon
,
Moumita
Bhattacharya
,
Robert
Baumgarten
,
Ryan T.
VanderLinden
and
Caroline T.
Saouma
*
,
Austin T.
Cannon
,
Moumita
Bhattacharya
,
Robert
Baumgarten
,
Ryan T.
VanderLinden
and
Caroline T.
Saouma
*
Department of Chemistry, University of Utah, 315 S. 1400 E., Salt Lake City, UT 84112, USA. E-mail: caroline.saouma@utah.edu
First published on 21st September 2020
Abstract
A cobalt(I) complex is shown to be capable of both electrocatalytic reduction and hydrogenation of CO2 to formate. Several proposed intermediates are characterized and thus form the basis for a proposed mechanism that allows for the dual reactivity: reduction of CO2via H2 addition, and H+/e− equivalents. The work makes use of a novel tris(phosphino) ligand. When a pendent amine is attached to the ligand, no change in catalytic reactivity is observed.
Reduction of CO2 to fuels and/or fuel precursors is integral to minimize global warming and advance future energy schemes.1 One approach is to use H2 to hydrogenate CO2 to formic acid (FA) or MeOH, though challenges include transportation of the gas, the necessity of high pressures of H2 and/or elevated temperatures required for many catalysts, and improving catalyst performance. Lifecycle analysis for CO2 hydrogenation to FA (using a homogeneous catalyst) suggests that this approach can decrease the net greenhouse gas emissions when compared to FA production from CO.2 Despite showing an improvement, this analysis also indicates that H2 production accounts for a significant amount of the emissions.3
An alternative method is the solar-derived electrochemical reduction of CO2.4 While the 2e−/2H+ reduction of CO2 to CO is well-established, reduction to FA has proven more challenging at homogenous systems due to competing H2 production.5 This latter reaction can be thought of as an electrochemical hydrogenation. It necessitates a proton source capable of generating a metal hydride, and that the subsequent insertion of CO2 be favoured over loss of H2; both reactions have similar thermodynamic driving forces.4 Berben's group showed that selectivity for FA over H2 can be achieved by exclusion of a pendent proton shuttle, which alters the kinetics of proton transfer to the active-site.6 Recently, the groups of Kubiak7 and Yang5 have shown how H2 production can be circumvented on thermodynamic arguments if the product is formate and not FA.
Given the widespread utility of hydrogenations, advancement of electrochemical alternatives may have significant impact. Waymouth showed that a Ru transfer hydrogenation catalyst can serve as an electrocatalyst for the oxidation of alcohols to ketones;8 in this system a cationic solvent species is proposed as an intermediate. With regards to CO2 conversion to formate, Meyer and Brookhart reported that the 2e−/1H+ reduction of a PCP-ligated IrH(MeCN)2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 gives a species capable of inserting CO2, with subsequent formate release. This system also necessitates a labile solvent molecule to avoid an 18-electron species that cannot be reduced. The limited literature examples of electrocatalytic hydrogenations re-enforce the need for better understanding how the two mechanistic pathways intersect, as well as establishing catalyst design criteria that allows for the desired reactivity.
9 gives a species capable of inserting CO2, with subsequent formate release. This system also necessitates a labile solvent molecule to avoid an 18-electron species that cannot be reduced. The limited literature examples of electrocatalytic hydrogenations re-enforce the need for better understanding how the two mechanistic pathways intersect, as well as establishing catalyst design criteria that allows for the desired reactivity.
Herein we describe a new family of Co complexes that perform both hydrogenation and electrocatalytic reduction of CO2 to formate with excellent product selectivity. To our knowledge, this is the first system capable of this dual reactivity: reaction of CO2 with H2 to give FA, and reaction of CO2 with H+ and e− equivalents to selectively give FA (over competing H2 production). Mechanistic studies indicate how the mechanisms are related. The ligand features a pendent amine that does not impact either reaction type.
To explore the dual reactivity of electrocatalytic reduction10,11 and hydrogenation12–15 of CO2, phosphine-ligated Co complexes were targeted. Electrocatalytic generation of Co–H is known to occur for proton reduction catalysts;16 one example17 employs a tris(phosphino) ligand that has also been shown to catalytically hydrogenate CO2 to MeOH using Co.18 To explore the role that pendent proton-relays may have on both catalytic pathways, a tris(phosphino) scaffold was developed that features a single pendent amine and is flexible in mer/fac coordination to the metal.
Tris(phosphino) ligands with a central phosphine that can be functionalized were prepared (Chart 1). The ligands that feature a pendent amine are readily prepared by addition of a suitable amine and paraformaldehyde to the precursor secondary phosphine (see ESI†).19 For this study, two tertiary amines (Bz2NP3, Ph2NP3) were chosen for the pendent amine. A ligand with no amine, MeP3 was also synthesized.20
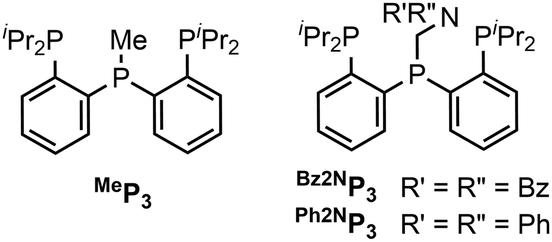 | ||
| Chart 1 Ligands and abbreviations used in this study. | ||
Metalation of the ligands is achieved by stirring equimolar ligand with CoCl2 or Co(PPh3)3Cl to give (RP3)CoCl2 or (RP3)CoCl respectively. Solid-state structures of (Bz2NP3)CoCl2, (Ph2NP3)CoCl2, (Bz2NP3)CoCl, (Ph2NP3)CoCl and (MeP3)CoCl were obtained and exemplary structures shown in Fig. 1. All of the Co(II) species feature two inner-sphere chloride ions, and have distorted square pyramidal geometry (τ ∼ 0.15).21 The Co(I) species are 4-coordinate and are best described as distorted tetrahedral (τ ∼ 0.75).22 In no instances does the amine nitrogen coordinate the metal centre.
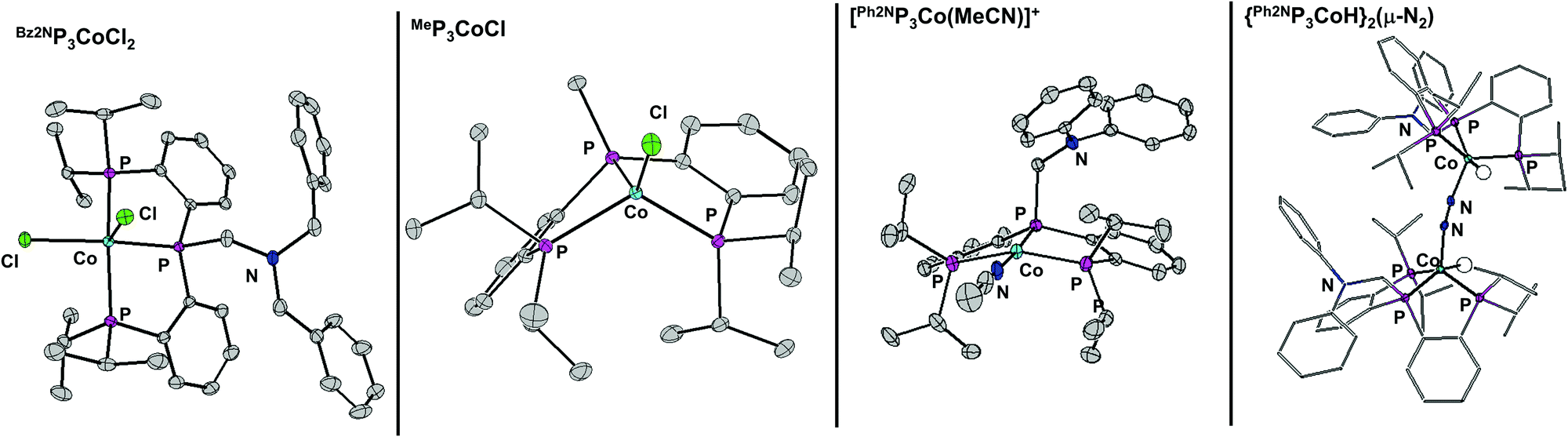 | ||
| Fig. 1 50% thermal ellipsoid plot of several complexes. All hydrogen atoms not located in the difference map are removed for clarity. Only the cation of [(Ph2NP3Co(MeCN))][BArF] is shown. | ||
Hydrogenation of CO2 under basic conditions was then explored with the Co(I) complexes (Table 1). No MeOH was observed by GC analysis, and the only product detected was formate.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Equiv. of base | Formate TONc |
| a Reactions run in 3 mL THF, at 150 °C for 20 h. b Reactions run in 10 mL THF with the conditions provided in the scheme. c Average of two runs with standard deviation in parenthesis. If no standard deviation, single run. d Run for 20 h. | ||||
| 1a | (Ph2NP3)CoCl | K3PO4 | 100 | 12 |
| 2a | (Ph2NP3)CoCl | KOtBu | 100 | 65 (±6) |
| 3a | (Ph2NP3)CoCl | DBU | 100 | 51 |
| 4b | [(Ph2NP3)Co(MeCN)][BArF] | KOtBu | 100 | 111 (±4) |
| 5b | (Bz2NP3)CoCl | KOtBu | 100 | 124 (±9) |
| 6b | (Bz2NP3)CoCl | KOtBu | 200 | 37 (±8) |
| 7b | (Bz2NP3)CoCl | KOtBu | 2000 | 242 |
| 8b | (MeP3)CoCl | KOtBu | 100 | 122 (±6) |
| 9b | (Bz2NP3)CoCl | — | — | 9 |
| 10b,d | — | KOtBu | 100 | 20 |
Entries 1–3 of Table 1 indicate that the base strength impacts catalysis; increasing the base strength from K3PO4 to KOtBu gives higher turnover number (TON), suggesting that a deprotonation event may limit the catalysis. Two of the Co(I)Cl species gave ∼quantitative TON with respect to base (entries 5, 8) when 100 eq. KOtBU is employed. Increasing the amount of base diminishes catalysis (entries 5–7); a color change is noted when large amounts of base are added to the catalyst solution, suggestive of catalyst degradation. Recycling studies indicate that a viable catalyst is present at the end of catalysis, though the paramagnetic nature of the complexes makes it difficult to ascertain the identity (see ESI†).
To determine if the chloride is pertinent to catalysis, Ph2NP3CoCl was treated with NaBArF to give cationic [(Ph2NP3Co(MeCN))][BArF] (BArF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate), the structure of which is shown in Fig. 1. Using the cation as a catalyst for the hydrogenation of CO2 improves the TON compared to that of the chloride (entries 2 and 4) and indicates that ∼quantitative conversion is possible using complexes of all ligands examined.
A mechanism that includes CO2 insertion into a Co–H (to give Co–OCHO) seems plausible and hence this reactivity was explored. Addition of 2 eq. of NaBHEt3 to a stirring THF solution of Ph2NP3CoCl2 at −70 °C results in formation of a new species. The 31P NMR spectrum shows two singlets at 101.6 and 98.9 ppm, suggesting that a single diamagnetic Co(I) species has formed. The corresponding 1H NMR spectrum shows a doublet of triplet at −11.35 ppm, consistent with a Co–H and IR analysis shows a stretch at 2082 cm−1. Vapor diffusion of benzene into heptane gave crystals suitable for diffraction, and the solid-state structure indicates the formation of a dimeric species, {Ph2NP3CoH}2(μ-N2) (Fig. 1). Each Co is 5-coordinate with the hydride in the plane of the three phosphines and the N2 coordinating in the apical position.
To determine if the hydride is sufficiently hydridic to insert CO2, 0.85 atm of CO2 was added to a solution of {Ph2NP3CoH}2(μ-N2). NMR analysis shows complete conversion of the diamagnetic hydride to a new paramagnetic species. Now, the IR spectrum shows disappearance of the hydride resonance and a new peak at 1628 cm−1, consistent with formation of a species such as Ph2NP3Co-OCHO. The related species, (PPh3)3Co(H)(N2), inserts CO2 to give (PPh3)3Co(OCHO),23 and the corresponding formate stretch is at 1620 cm−1.
A proposed mechanism is shown in Scheme 1. Entry into the catalytic cycle occurs from the reaction of RP3CoCl with H2 and base, which would give a 5-coordinate species such as RP3Co(H)(L) (L = N2 or solvent). Subsequent insertion of CO2 gives RP3Co-OCHO. The resulting 16-electron species RP3Co-OCHO may then coordinate H2 to give the proposed 18-electron RP3Co(H2)-OCHO. Base-mediated deprotonation of the bound H2 coupled with formate loss regenerates RP3Co(H)(L). Another mechanism would be deprotonation of the bound H2 by the inner-sphere formate to generate RP3Co(H)(L) and formic acid (B = formate in Scheme 1). Indeed, this may explain the >100 TON (entries 4, 5, 8) when only 100 equivalents of base is added, as well as the low TON obtained in the absence of base (entry 9). This is proposed to be a minor pathway that is viable in the absence of base, that proceeds with slower kinetics.
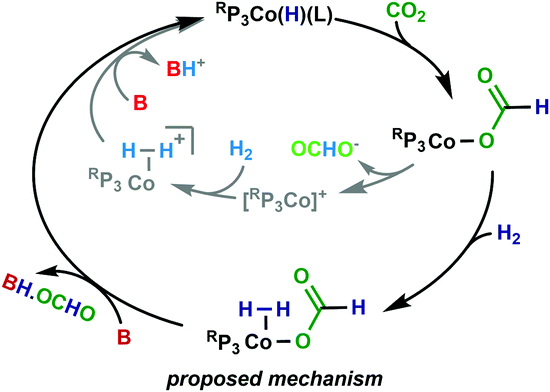 | ||
| Scheme 1 Proposed mechanism for hydrogenation of CO2 to formate, with alternate mechanism shown in grey. | ||
An alternative mechanism that has been proposed for related complexes is that the formate dissociates from the Co to give [Ph2NP3Co(MeCN)]+ and free formate (Scheme 1, inner pathway). The cation then coordinates H2 and base-mediated deprotonation of the bound H212 or oxidative addition product (not shown) then ensues.13,14 Given that both [Ph2NP3Co(MeCN)]+ and RP3Co-OCHO are stable, this seems unlikely. Moreover, in related work on Ru we have shown that binding of formate to a cationic Ru centre is favorable.24
Though it was envisioned that the pendent amine may facilitate deprotonation of H2via hydrogen-bonding,25 the similar catalytic performances amongst all the catalysts indicates that such an effect, if present, is irrelevant in the catalytic hydrogenations investigated.
With the feasibility to hydrogenate CO2 to formate established, we sought to determine if electrocatalytic hydrogenation of CO2 is also accessible. The cyclic voltammograms (CVs) of several complexes are shown in Fig. 2. Cationic [Ph2NP3Co(MeCN)]+ shows a reversible reduction at −0.863 V (vs. Fc+/0) that corresponds to the Co(II/I) couple. A second reduction event occurs at ∼−1.9 V, with a return oxidation at ∼−1.3 V. This tentatively is assigned to the Co(I/0) couple. Consistent with this, the reaction of Ph2NP3CoCl with Na/Hg gives Ph2NP3Co(N2) (see ESI†); the irreversible nature of the couple is attributed to N2 coordination upon reduction. The irreversibility may also be attributed to different numbers and types of L-type ligands upon reduction (L = MeCN or N2). The CVs of Ph2NP3CoCl2 and Ph2NP3CoCl are similar, and show a quasi-reversible reduction at −1.050 V. Both CVs show peaks that correspond to [Ph2NP3Co(MeCN)]+, consistent with chloride loss upon reduction to Co(0). Notably, the peaks that correspond to the second reduction are super-imposable with those in the CV of [(Ph2NP3Co(MeCN))]+. Modest changes in the reduction potentials is anticipated as the R group on the central phosphine is varied.10 Indeed, the Co(II/I) potential of Bz2NP3CoCl occurring at −1.013 V, and that of MeP3CoCl at −1.089 V.
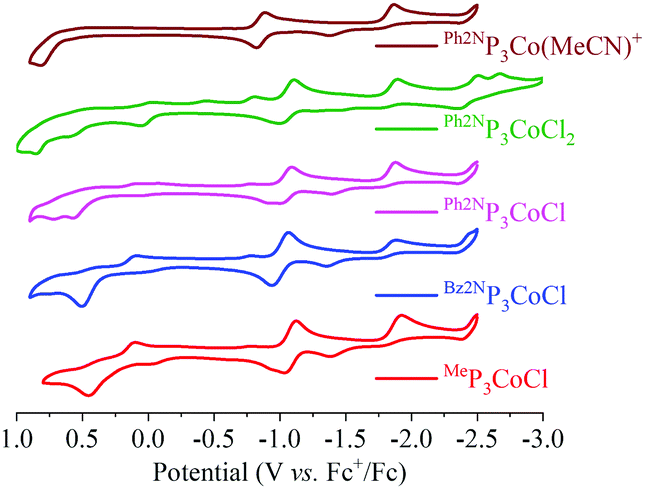 | ||
| Fig. 2 Cyclic voltammograms of the various Co complexes. Conditions: GC working electrode, 0.1 M TBAPF6 electrolyte in MeCN. Initial scan is in the positive direction. | ||
In the presence of 50 eq. water, no noticeable changes to the CVs are observed for all RP3CoCl (Fig. 3 and ESI†). However, upon addition of CO2, a catalytic current is observed, suggesting formation of CO or formate. The current increases further with 1617 eq. H2O (3% by volume, see ESI†) at potentials close to the CoI/0 couple; other Co electrocatalysts reduce CO2 at potentials well shifted from the redox couples of the catalyst.10 Indeed, rapid current enhancement at ∼−2.5 V suggests that there may be two pathways for catalytic reduction.
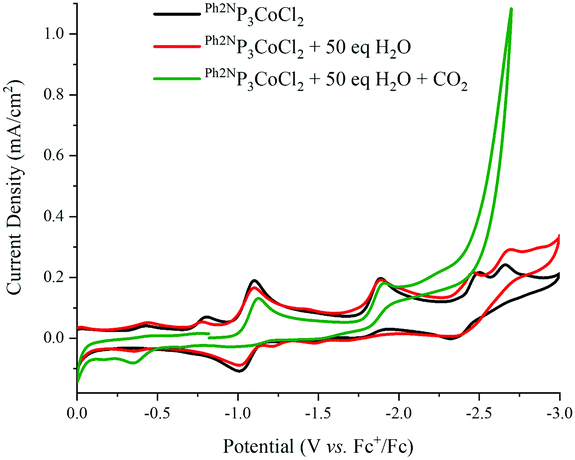 | ||
| Fig. 3 Cyclic voltammograms of 1 mM Ph2NP3CoCl2 under various conditions. (Black): under N2; (red): in the presence of 50 eq. H2O; (green): in the presence of 50 eq. H2O and CO2. Conditions: GC working electrode, 0.1 M TBAPF6 electrolyte in MeCN solvent, scan rate of 0.06 V s−1, initial scan is positive. | ||
Controlled potential electrolysis with 3% water and 0.85 atm of CO2 at −2.1 V vs. Fc/Fc+ was conducted using MeP3CoCl and Bz2NP3CoCl as the catalyst (Table 2). This potential is chosen to see if catalysis does occur near the reduction potential of the complexes. In both instances, no CO was detected in the headspace, and H2 is only produced in small quantities with both catalysts. No other gaseous products are produced, and the solution phase shows the presence of formate and MeOH.
The catalysts are stable, as ascertained by CVs after electrolysis and the steady current (see ESI†).
A proposed mechanism is shown in Scheme 2. Upon reduction to RP3Co0L (L = N2 or solvent), protonation ensues to give [RP3CoIIH]+. This is reduced at the electrode to give RP3CoIH, which then inserts CO2 to give RP3CoI(OCHO); this sequence being identical to that in the hydrogenation reaction. Reduction to RP3Co0(OCHO) and subsequent formate release then regenerates RP3Co0L. The lack of lability of the formate in RP3CoI(OCHO) and the cathodic potential of catalysis is consistent with this EC mechanism. While we cannot rule out initial CO2 insertion to [RP3CoIIH]+ followed by reduction, this reactivity is not known for this system. Finally, the pH of the solution increases during the course of catalysis, indicating that formate and not formic acid is lost.
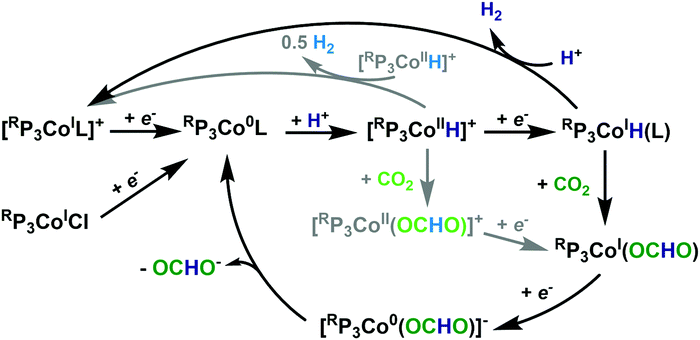 | ||
| Scheme 2 Proposed (black) & alternative (gray) mechanisms for CO2 and H+ reduction. | ||
Regarding proton reduction, the following can be gleaned. As RP3CoIH is stable, we rule out a bimetallic mechanism that would generate RP3Co0L. A bimetallic mechanism from [RP3CoII-H]+ also seems unlikely, as it would not explain why H2 is only produced in the presence of pendent amines; these species are also more sterically encumbering and hence should minimize this pathway on steric grounds. Protonation of RP3CoIH seems most plausible, and literature precedence is consistent with proton relays enhancing H2 production over formate.6
The work presented here provides the first example of a well-defined catalyst that can hydrogenate CO2 to formate and electrocatalytically reduce CO2 to formate. Notably, the latter reaction occurs with good selectivity for formate. The stability of several intermediates, including a cationic solvent species that seems essential for the dual reactivity, allows for further mechanistic insight. Optimization studies and detailed mechanistic work is ongoing.
The authors gratefully acknowledge the NSF CAREER (1945646), NSF REU (1659579) and start-up funds. NIH provided funds for the NMR facilities (1S10OD25241-01, 1C06RR017539-01A1, 3R01GM063540-17W1). We acknowledge Dr. Stacey J. Smith and BYU for the use of their single crystal X-ray instrumentation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS.
- A. Sternberg, C. M. Jens and A. Bardow, Green Chem., 2017, 19, 2244–2259 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS.
- B. M. Ceballos and J. Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12686–12691 CrossRef CAS.
- N. D. Loewen, E. J. Thompson, M. Kagan, C. L. Banales, T. W. Myers, J. C. Fettinger and L. A. Berben, Chem. Sci., 2016, 7, 2728–2735 RSC.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- K. M. Waldie, K. R. Flajslik, E. McLoughlin, C. E. D. Chidsey and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 738–748 CrossRef CAS.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS.
- F.-W. Liu, J. Bi, Y. Sun, S. Luo and P. Kang, ChemSusChem, 2018, 11, 1656–1663 CrossRef CAS.
- A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS.
- M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS.
- E. S. Rountree, D. J. Martin, B. D. McCarthy and J. L. Dempsey, ACS Catal., 2016, 6, 3326–3335 CrossRef CAS.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS.
- E. Payet, A. Auffrant, X. F. Le Goff and P. L. Floch, J. Organomet. Chem., 2010, 695, 1499–1506 CrossRef CAS.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356, 10.1039/DT9840001349.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964, 10.1039/B617136B.
- L. S. Pu, A. Yamamoto and S. Ikeda, J. Am. Chem. Soc., 1968, 90, 3896 CrossRef CAS.
- C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS.
- M. Rakowski Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2010200–2010206, 2022544 and 2022545. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04310a |
| This journal is © The Royal Society of Chemistry 2020 |