
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Ar@C60 using molecular surgery†
Sally
Bloodworth
 ,
Gabriela
Hoffman
,
Mark C.
Walkey
,
George R.
Bacanu
,
Julie M.
Herniman
,
Malcolm H.
Levitt
and
Richard J.
Whitby
*
,
Gabriela
Hoffman
,
Mark C.
Walkey
,
George R.
Bacanu
,
Julie M.
Herniman
,
Malcolm H.
Levitt
and
Richard J.
Whitby
*
Chemistry, Faculty of Engineering and Physical Sciences, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: rjw1@soton.ac.uk
First published on 4th August 2020
Abstract
Synthesis of Ar@C60 is described, using a route in which high-pressure argon filling of an open-fullerene and photochemical desulfinylation are the key steps for >95% encapsulation of the noble gas. Enrichment by recycling HPLC leads to quantitative incorporation of argon in the product endofullerene, with a mass recovery of tens of milligrams, allowing the first characterisation of fine structure in the solution 13C NMR spectrum.
Endohedral fullerenes (endofullerenes) are substances whose molecules consist of closed fullerene cages (for example C60 or C70), each encapsulating a single atom or small molecule.1 The C60 cavity is able to accommodate the noble gas atoms from helium to xenon,2,3 and the existence of a noble gas endofullerene was first demonstrated when encapsulation of 4He in C60 (denoted 4He@C60) was detected from gas-phase neutralisation of 4HeC60+˙, obtained by collision of accelerated C60+˙ with helium gas.4
Naturally occurring noble gas endofullerenes of extraterrestrial origin have since been detected in meteor deposits.5 They are materials of great interest for study of their non-bonding intermolecular interactions and the quantum energy level structure of the isolated atom. Encapsulation of the larger noble gas atoms argon, krypton, and xenon leads to electronic and structural distortions of the C60 cage – determined by powder diffraction,6 NMR,7 IR, Raman and X-ray spectroscopy,8 variance in the critical temperature of superconducting alkali-doped endofullerenes,9 exohedral reactivity,7 and theoretical studies.8a,10,11 Samples of Ar@C60, Kr@C60 and Xe@C60 are, so far, obtained from C60 by direct encapsulation of the noble gas atom under high temperature and pressure, and typically only 0.2–0.4% incorporation is possible using this method.3,12 Subsequent removal of empty C60 using preparative, recycling HPLC leads to enrichment of the sample (for example, approx. 1.3 mg of Ar@C60 has been prepared with >98% purity in several batches with overall yield of <0.1% from C60).6,9 Although evidence for improved 18% direct encapsulation of argon by laser-vaporisation of C60 in the presence of the gas has been reported,13 there remains no synthetic method to obtain more than ∼1 mg of pure Ar@C60, Kr@C60 or Xe@C60.
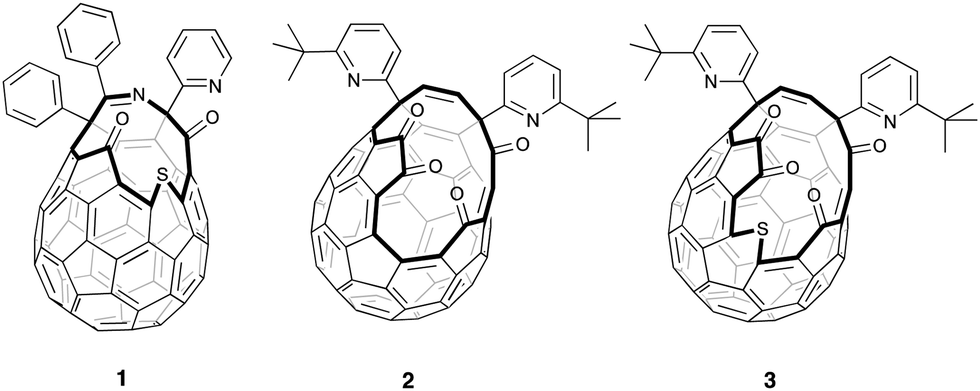 | ||
| Fig. 1 Open-cage fullerenes 1–3 are key intermediates in the synthesis of He@C60 and H2@C60 (from 1); H2O@C60, H2@C60 and HF@C60 (from 2); and CH4@C60 (from 3). | ||
In contrast, larger scale synthesis of 4He@C60 (38 mg with 30% 4He filling, enriched to a sample with >95% filling) has been achieved by Komatsu and Murata using multi-step ‘molecular surgery’, in which an atom or small molecule enters the cavity of an open-cage fullerene whose opening is then sutured to restore the carbon cage around the trapped endohedral species. In their pioneering work, synthesis of H2@C60 as well as 4He@C60 was accomplished from open fullerene 1, by insertion of H2 directly into 1 and of 4He into the sulfoxide derivative, under high pressure (Fig. 1).14 The same authors also pioneered the synthesis and orifice-suture of open-cage fullerene 2 in the first reported synthesis of H2O@C60,15 and we have adapted these procedures for preparation of H2@C60,16 and HF@C6017 from 2.
The orifice of 2 is too small to accommodate argon, but the bigger opening of fullerene 3 permits entry of large guests including methane,18 and we recently described >95% filling of 3 with CH4 under >1500 atm of methane at 190 °C, followed by closure of the orifice of CH4@3 to obtain CH4@C60.19 Herein, we report that high encapsulation of argon by 3 can be achieved under similar conditions, and closure allows synthesis of Ar@C60 with a step-change improvement in argon incorporation, mass recovery and yield when compared to the direct encapsulation method.
Synthesis of Ar@C60 was carried out according to the procedures shown in Scheme 1. Fullerene 3 was prepared according to the method reported by Murata, by insertion of sulfur into 2 in the presence of tetrakis(dimethylamino)ethylene.20 In order to estimate conditions for argon filling, the activation energy for entry of argon into the cavity of 3 was calculated by density functional theory21 and compared with that of methane. We found a lower barrier to argon entry (ΔH‡entry(Ar) = 55 kJ mol−1; ΔH‡entry(CH4) = 86 kJ mol−1) with similar binding enthalpies (ΔHbind(Ar) = −46 kJ mol−1; ΔHbind(CH4) = −50 kJ mol−1) and anticipated quantitative incorporation of argon under conditions similar to those which gave >95% CH4 encapsulation. Accordingly, heating powdered 3 at 180 °C under approx. 1400 atm of argon for 17.5 h22 gave quantitative recovery of Ar@3, with argon filling of >99% estimated from the 1H NMR and positive ion ESI mass spectra. Empty open-fullerene 3 is known to rapidly take up water at room temperature23,24 (endohedral H2O δH = −11.50 ppm in 3, 300 MHz, CDCl3)20 yet no resonance corresponding to endohedral water is seen in the 1H NMR spectrum of Ar@3 in wet CDCl3. Furthermore, no peaks corresponding to empty 3, or H2O@3, were observed in the high resolution ESI+ mass spectrum of Ar@3 (ESI†).
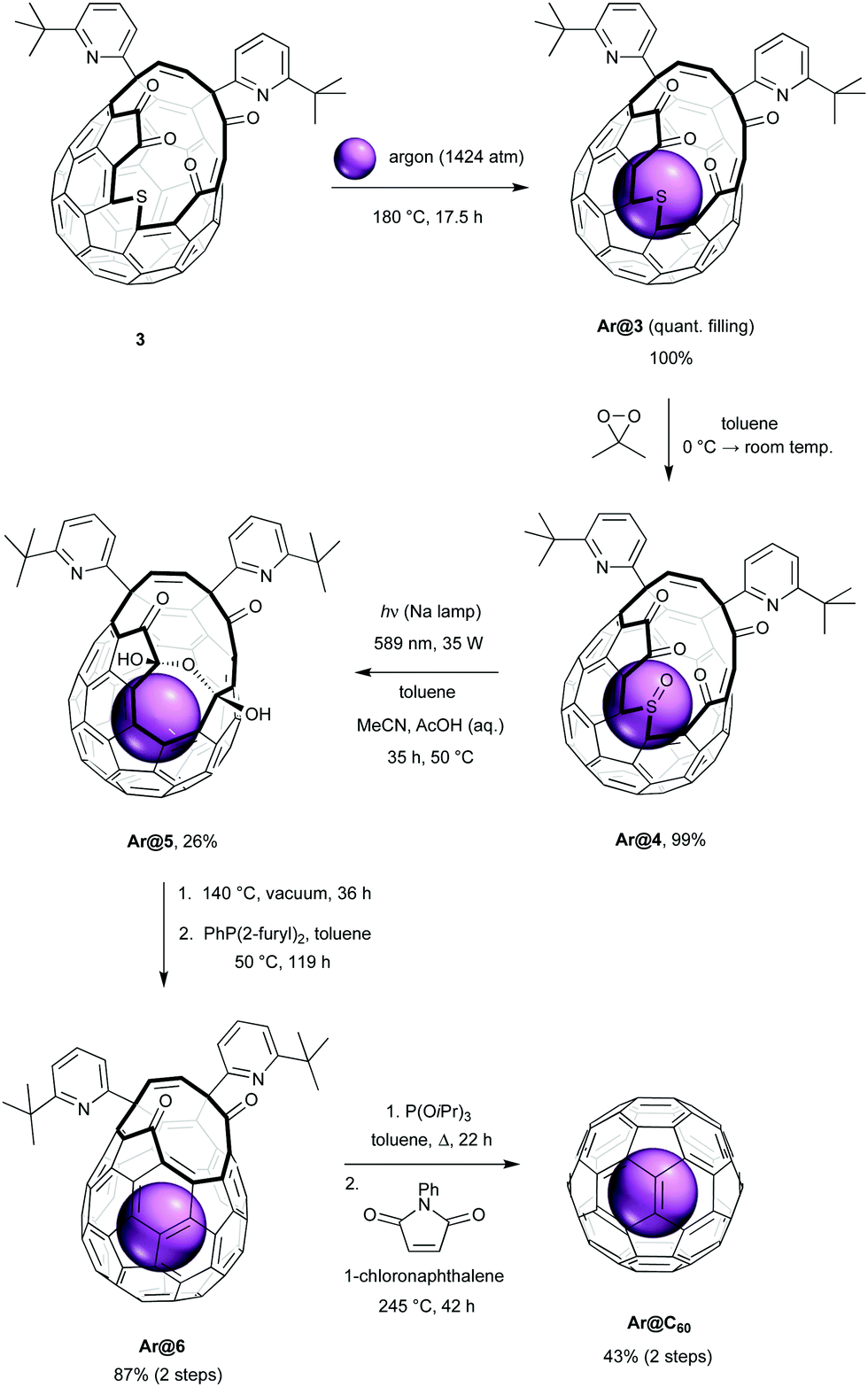 | ||
| Scheme 1 Synthesis of Ar@C60. | ||
Clean and high-yielding oxidation of Ar@3 with dimethyldioxirane gave the sulfoxide Ar@4, and we now applied conditions for photochemical desulfinylation of Ar@4 that were developed for our synthesis of CH4@C60.19 Desulfinylation is the first of the ‘closure’ steps that suture the orifice of the open-cage fullerene and a similar photochemical ring-contraction by removal of the sulfinyl group (SO) of other sulfoxide open-cage endofullerenes has been reported, using visible-light irradiation in toluene or benzene.14,25 Photochemical desulfinylative ring-contraction of sulfoxide 4 re-forms fullerene 2 which is unstable under the reaction conditions, but using a vigorously stirred two-phase mixed solvent system of toluene, acetonitrile and acetic acid (10% v/v aqueous) allows in situ trapping of 2 as the more stable bis(hemiketal) 5. Upon irradiation of Ar@4 for 35 h under these conditions, at 50 °C using a low-pressure sodium lamp, the bis(hemiketal) Ar@5 was obtained in 26% yield of isolated product. Our previously reported photochemical desulfinylation of CH4@4 led to the corresponding bis(hemiketal) CH4@5 in only 13% yield under the same conditions,19 indicating that contraction of the cage-opening is less inhibited by argon, the smaller endohedral species.
Under the mixed (part aqueous) conditions of the photochemical desulfinylation step, the empty open fullerene 4 present in <1% is expected to encapsulate water. In order to avoid contamination of the eventual Ar@C60 product with trace H2O@C60, Ar@5 was now heated at 140 °C under dynamic vacuum for 36 h. This achieves both dehydration of the bis(hemiketal) to form Ar@2, and removal of any endohedral water from the small portion of the material that is not argon-filled. Reduction of Ar@2 using di-(2-furyl)phenylphosphine was next carried out at 50 °C, a temperature too low for re-entry of water to occur, to give Ar@6 in 87% yield. Argon filling of >95% was estimated from the high resolution ESI+ mass spectrum of Ar@6 by comparison of peak intensity for the filled and empty species, m/z = 1111.1691, [12C82H27ArN2O2]+ and m/z = 1071.2081, [12C82H27N2O2]+. No evidence of H2O@6 was found. We assume that the lower argon filling of >95% in Ar@6 (cf. >99% for Ar@4) and presence of 3–4% of empty 6 arises from some escape of argon from 4 under the conditions of the desulfinylation, something we did not observe for CH4@4, and reflects the calculated lower barrier to argon loss from 4 (ΔH‡exit(Ar) = 110 kJ mol−1; ΔH‡exit(CH4) = 150 kJ mol−1). Loss of argon from 2 in the dehydration step is unlikely (ΔH‡exit(Ar) = 216 kJ mol−1; ΔH‡exit(H2O) = 100 kJ mol−1).21
Finally, the opening of Ar@6 was closed under conditions previously reported for the synthesis of H2O@C60,16 and Ar@C60 was obtained with 94.7% filling. Removal of the 5.3% contaminant C60 was achieved by recycling preparative HPLC,6,9,26 to give Ar@C60 with 100% incorporation of the noble gas. In this way, we were able to prepare 20 mg of pure Ar@C60 in 9.6% yield from fullerene 3. In our hands, fullerene 3 is obtained from C60 in 29–30% using the reported procedures,15,20,27 so an overall yield of >2.7% Ar@C60 is achieved.
The positive-ion atmospheric pressure photoionization (APPI) mass spectrum of Ar@C60 is in agreement with the calculated isotope pattern (Fig. 2).
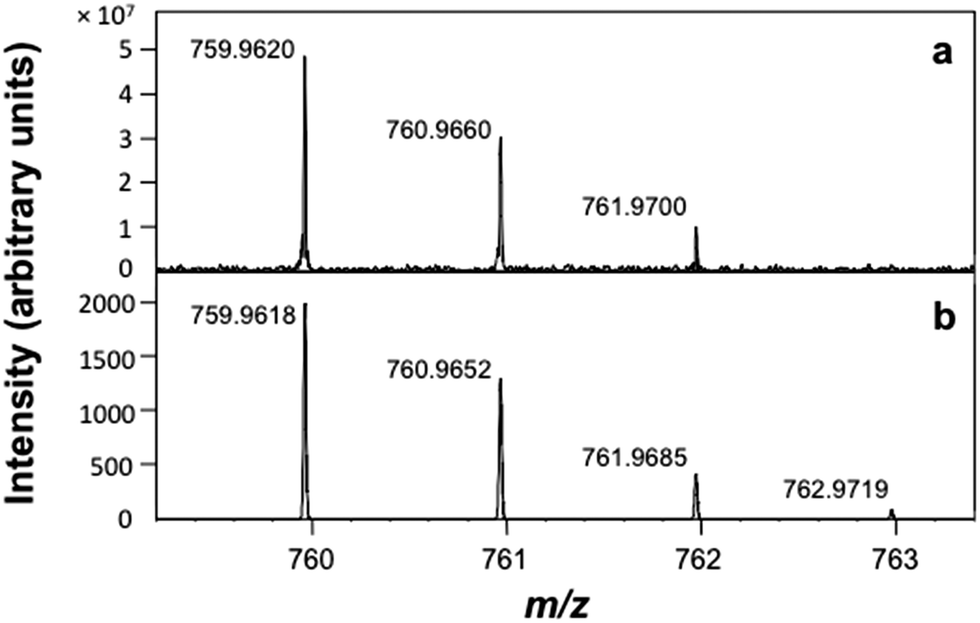 | ||
| Fig. 2 Positive-ion APPI mass spectrum of Ar@C60. (a) Experimental data and (b) calculated isotope pattern, error −0.2 ppm. | ||
The 13C NMR resonance of Ar@C60 was measured with a chemical shift of δC = 142.98 ppm in 1,2-dichlorobenzene-d4 at 298 K, deshielded by Δδ = +0.18 ppm relative to empty C60 (δC = 142.80 ppm). Dragoe and colleagues have reported a comparable value in benzene-d6 of Δδ = +0.17 ppm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,9 and, in the noble gas@C60 series, deshielding of the cage 13C NMR resonance with respect to empty C60 increases with the van der Waals radius of the enclosed atom: He@C60, Δδ = +0.02 ppm;14b Ar@C60, Δδ = +0.18 ppm; Kr@C60, Δδ = +0.39 ppm;12d Xe@C60, Δδ = +0.96 ppm.28 A pair of small side peaks with intensity ratio 2:1 is also observed in the 13C NMR spectrum, shifted by −12.50 ppb and −19.86 ppb with respect to the main fullerene resonance (Fig. 3). These peaks are assigned to minor isotopomers of Ar@C60 that contain a pair of neighbouring 13C nuclei separated by one bond, and two peaks are observed since there are two types of carbon–carbon bond in C60, either a hexagon–pentagon (HP) or shorter hexagon–hexagon (HH) shared edge, present in a 2:1 ratio respectively. The shifts of the side peaks relative to the main peak correspond to one-bond secondary isotope shifts (1ΔC = 12.50 ± 0.01 ppb for the inner HP peak, and 1ΔC = 19.86 ± 0.02 ppb for the outer HH peak). These values are slightly smaller in magnitude than those found for empty C60 (1ΔC = 12.56 ± 0.01 ppb and 1ΔC = 19.98 ± 0.02 ppb for HP and HH peaks respectively) and lie between the values observed for H2@C60 and H2O@C60.29
6,9 and, in the noble gas@C60 series, deshielding of the cage 13C NMR resonance with respect to empty C60 increases with the van der Waals radius of the enclosed atom: He@C60, Δδ = +0.02 ppm;14b Ar@C60, Δδ = +0.18 ppm; Kr@C60, Δδ = +0.39 ppm;12d Xe@C60, Δδ = +0.96 ppm.28 A pair of small side peaks with intensity ratio 2:1 is also observed in the 13C NMR spectrum, shifted by −12.50 ppb and −19.86 ppb with respect to the main fullerene resonance (Fig. 3). These peaks are assigned to minor isotopomers of Ar@C60 that contain a pair of neighbouring 13C nuclei separated by one bond, and two peaks are observed since there are two types of carbon–carbon bond in C60, either a hexagon–pentagon (HP) or shorter hexagon–hexagon (HH) shared edge, present in a 2:1 ratio respectively. The shifts of the side peaks relative to the main peak correspond to one-bond secondary isotope shifts (1ΔC = 12.50 ± 0.01 ppb for the inner HP peak, and 1ΔC = 19.86 ± 0.02 ppb for the outer HH peak). These values are slightly smaller in magnitude than those found for empty C60 (1ΔC = 12.56 ± 0.01 ppb and 1ΔC = 19.98 ± 0.02 ppb for HP and HH peaks respectively) and lie between the values observed for H2@C60 and H2O@C60.29
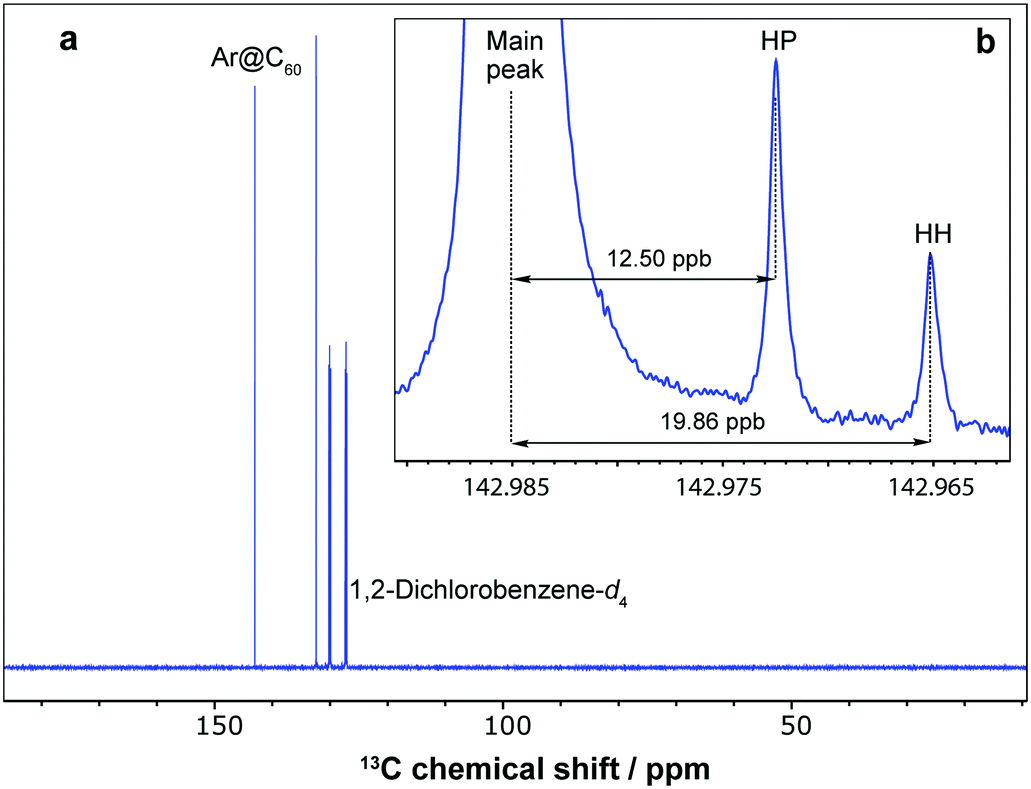 | ||
| Fig. 3 (a) 13C NMR spectrum of Ar@C60 (100% argon filling), 24 mM solution in degassed 1,2-dichlorobenzene-d4 at a field of 176 MHz and 298 K, acquired with 32 transients. (b) Expanded view of the base of the Ar@C60 resonance, acquired at 298 K with 848 transients, to show side peaks arising from minor isotopomers with two adjacent 13C nuclei that share either a hexagon–pentagon (HP) or hexagon–hexagon (HH) edge. | ||
The room temperature infrared and UV spectra of Ar@C60 showed no changes in the positions of peaks, compared with C60.
In conclusion we have reported the first synthesis of Ar@C60 using molecular surgery, in which high-pressure filling of an open-fullerene and photochemical desulfinylation are the key steps for >95% encapsulation of the noble gas. Further enrichment by recycling HPLC has enabled recovery of the product with quantitative incorporation of endohedral argon, on a scale of tens of milligrams. Our method overcomes the limitation of low mass recovery in the earlier direct encapsulation method and will allow measurement of the energies of the quantised translational modes of the argon atom using IR, THz and inelastic neutron scattering spectroscopy – a sensitive test of current theoretical models of dispersive interactions. The availability of Ar@C60 will also facilitate studies of the influence of the endohedral atom upon exohedral reactivity of the fullerene.
The authors thank Prof. G. John Langley for access to APPI mass spectrometry, and acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton. This research was supported by EPSRC (UK) (EP/M001962/1 and EP/P009980/1) including core capability (EP/K039466), and the European Research Council (786707 – FunMagResBeacons).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews see: (a) R. G. Lawler, Nanostruct. Sci. Technol., 2017, 229 CrossRef; (b) M. H. Levitt, Philos. Trans. R. Soc., A, 2013, 371, 20120429 CrossRef PubMed; (c) A. A. Popov, S. F. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989 CrossRef CAS PubMed.
- H. A. Jiménez-Vázquez and R. J. Cross, J. Chem. Phys., 1996, 104, 5589 CrossRef.
- M. Saunders, H. A. Jiménez-Vázquez and R. J. Cross, J. Am. Chem. Soc., 1994, 116, 2193 CrossRef CAS.
- T. Weiske, T. Wong, W. Krätschmer, J. K. Terlouw and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1992, 31, 183 CrossRef.
- L. Becker, R. J. Poreda and T. E. Bunch, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2979 CrossRef CAS.
- A. Takeda, Y. Yokoyama, S. Ito, T. Miyazaki, H. Shimotani, K. Yakigaya, T. Kakiuchi, H. Sawa, H. Takagi, K. Kitazawa and N. Dragoe, Chem. Commun., 2006, 912 RSC.
- M. Frunzi, R. J. Cross and M. Saunders, J. Am. Chem. Soc., 2007, 129, 13343 CrossRef CAS PubMed.
- (a) F. Cimpoesu, S. Ito, H. Shimotani, H. Takagi and N. Dragoe, Phys. Chem. Chem. Phys., 2011, 13, 9609 RSC; (b) N. Dragoe, A. M. Flank, P. Lagarde, S. Ito, H. Shimotani and H. Takagi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 155448 CrossRef; (c) S. Ito, A. Takeda, T. Miyazaki, Y. Yokoyama, M. Saunders, R. J. Cross, H. Takagi, P. Berthet and N. Dragoe, J. Phys. Chem. B, 2004, 108, 3191 CrossRef CAS.
- K. Yakigaya, A. Takeda, Y. Yokoyama, S. Ito, T. Miyazaki, T. Suetsuna, H. Shimotani, T. Kakiuchi, H. Sawa, H. Takagi, K. Kitazawa and N. Dragoe, New J. Chem., 2007, 31, 973 RSC.
- R. B. Darzynkiewicz and G. E. Scuseria, J. Phys. Chem. A, 1997, 101, 7141 CrossRef CAS.
- (a) A. Bil and C. A. Morrison, J. Phys. Chem. A, 2012, 116, 3413 CrossRef CAS PubMed; (b) S. Osuna, M. Swart and M. Solà, Chem. – Eur. J., 2009, 15, 13111 CrossRef CAS PubMed; (c) S. Osuna, M. Swart and M. Solà, Phys. Chem. Chem. Phys., 2011, 13, 3585 RSC.
- (a) R. J. Cross, A. Khong and M. Saunders, J. Org. Chem., 2003, 68, 8281 CrossRef CAS PubMed; (b) M. Saunders, R. J. Cross, H. A. Jiménez-Vázquez, R. Shimshi and A. Khong, Science, 1996, 271, 1693 CrossRef CAS; (c) M. Saunders, H. A. Jiménez-Vázquez, R. J. Cross and R. J. Poreda, Science, 1993, 259, 1428 CrossRef CAS PubMed; (d) K. Yamamoto, M. Saunders, A. Khong, R. J. Cross, Jr., M. Grayson, M. L. Gross, A. F. Benedetto and R. B. Weisman, J. Am. Chem. Soc., 1999, 121, 1591 CrossRef CAS.
- B. Oksengorn, C. R. Acad. Sci. Paris, Serie IIc, Chimie, 2000, 3, 649 CAS.
- (a) K. Komatsu, M. Murata and Y. Murata, Science, 2005, 307, 238 CrossRef CAS PubMed; (b) Y. Morinaka, F. Tanabe, M. Murata, Y. Murata and K. Komatsu, Chem. Commun., 2010, 46, 4532 RSC; (c) M. Murata, Y. Murata and K. Komatsu, J. Am. Chem. Soc., 2006, 128, 8024 CrossRef CAS PubMed.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613 CrossRef CAS PubMed.
- A. Krachmalnicoff, M. H. Levitt and R. J. Whitby, Chem. Commun., 2014, 50, 13037 RSC.
- A. Krachmalnicoff, R. Bounds, S. Mamone, S. Alom, M. Concistrè, B. Meier, K. Kouřil, M. E. Light, M. R. Johnson, S. Rols, A. J. Horsewill, A. Shugai, U. Nagel, T. Rõõm, M. Carravetta, M. H. Levitt and R. J. Whitby, Nat. Chem., 2016, 8, 953 CrossRef CAS PubMed.
- S. Bloodworth, J. Gräsvik, S. Alom, K. Kouřil, S. J. Elliott, N. J. Wells, A. J. Horsewill, S. Mamone, M. Jiménez-Ruiz, S. Rols, U. Nagel, T. Rõõm, M. H. Levitt and R. J. Whitby, ChemPhysChem, 2018, 19, 266 CrossRef CAS.
- S. Bloodworth, G. Sitinova, S. Alom, S. Vidal, G. R. Bacanu, S. J. Elliott, M. E. Light, J. M. Herniman, G. J. Langley, M. H. Levitt and R. J. Whitby, Angew. Chem., Int. Ed., 2019, 58, 5038 CrossRef CAS.
- T. Futagoishi, M. Murata, A. Wakamiya, T. Sasamori and Y. Murata, Org. Lett., 2013, 15, 2750 CrossRef CAS PubMed.
- DFT calculations were carried out using M06-2X/cc-pVTZ//cc-pVDZ. For full details and references see the ESI†.
- Ar@3 was also recovered quantitatively with >99% filling after just 3.5 h, upon heating empty fullerene 3 at 190 °C under 1520 atm of argon.
- T. Futagoishi, M. Murata, A. Wakamiya and Y. Murata, Chem. Commun., 2017, 53, 1712 RSC.
- Y. Hashikawa, S. Hasegawa and Y. Murata, Chem. Commun., 2018, 54, 13686 RSC.
- M. Murata, S. Maeda, Y. Morinaka, Y. Murata and K. Komatsu, J. Am. Chem. Soc., 2008, 130, 15800 CrossRef CAS PubMed.
- B. A. DiCamillo, R. L. Hettich, G. Guiochon, R. N. Compton, M. Saunders, H. A. Jiménez-Vázquez, A. Khong and R. J. Cross, J. Phys. Chem., 1996, 100, 9197 CrossRef CAS.
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2017, 139, 16350 CrossRef CAS PubMed.
- M. S. Syamala, R. J. Cross and M. Saunders, J. Am. Chem. Soc., 2002, 124, 6216 CrossRef CAS.
- G. R. Bacanu, G. Hoffman, M. Amponsah, M. Concistrè, R. J. Whitby and M. H. Levitt, Phys. Chem. Chem. Phys., 2020, 22, 11850 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc04201c |
| This journal is © The Royal Society of Chemistry 2020 |