
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral molecular conductor with an insulator–metal transition close to room temperature†‡
Jonathan I.
Short
a,
Toby J.
Blundell
 a,
Sara J.
Krivickas
a,
Songjie
Yang
a,
John D.
Wallis
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b and
Lee
Martin
*a
a,
Sara J.
Krivickas
a,
Songjie
Yang
a,
John D.
Wallis
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b and
Lee
Martin
*a
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan
First published on 9th July 2020
Abstract
Materials exhibiting both chirality and conductivity do not exist in nature and very few examples have been synthesised. We report here the synthesis of a chiral molecular metal which remains metallic down to at least 4.2 K. This material also exhibits room-temperature switching capabilities with a transition upon cooling below 10 °C.
Bulk chiral molecular conductors are highly sought after because they can help formulate a theoretical model for electron conduction in chiral materials by exhibiting electrical magneto-chiral anisotropy (eMChA). The first observation of the effect of chirality upon superconductivity was recently observed in an individual nanotube of tungsten disulphide which shows asymmetric electron transport in an applied magnetic field.1 Differences in resistivity in applied magnetic fields between opposing enantiomers of the same material (eMChA) have been observed in bismuth helices,2 and carbon nanotubes.3 However, the lack of suitable enantiopure materials has made it difficult to formulate a theoretical model for electron conduction in bulk chiral conductors. The synthesis of radical-cation salts from enantiopure molecules (organosulfur donors, anions and/or solvents) provides an excellent opportunity to study eMChA in a chiral lattice which also shows conductivity or superconductivity. Radical-cation salts have been synthesized from a variety of chiral or racemic anions.4 The spatial arrangement of opposing Δ and Λ enantiomers of tris(oxalato)metallates in the anion layers determines the packing arrangement of the BEDT-TTFs in the donor layer which in turn determines the conducting behaviour.5 Using a chiral solvent for electrochemical growth of these salts can lead to a chiral guest molecule being included in the lattice,6 or to chiral induction producing a chiral salt which contains only a single enantiomer of tris(oxalato)metallate.7
The most successful method for producing chiral radical-cation salts has been by using enantiopure donors based on EDT-TTF8 or BEDT-TTF.9 Whilst still difficult to obtain highly crystalline materials, the donor tetramethyl-BEDT-TTF has produced several enantiopure salts.10 eMChA has been observed in salts of enantiopure DM-EDT-TTF with ClO4 which crystallise in enantiomorphic space groups P6222 and P6422, and show metallic behaviour above 40 K.11 Enantiopure DM-EDT-TTF also gives semiconducting salts with PF6 whilst the racemate adopts a different packing arrangement and shows metallic behaviour.12 Despite this seminal discovery, these are still the only materials of this type synthesised to date and more bulk chiral conducting materials are required to fully understand this phenomenon of eMChA.
We have previously reported the synthesis of the novel chiral donor molecule (1′R,5S)-N-(1′-phenylethyl)(BEDT-TTF)acetamide 1 (Scheme 1) along with its diastereomer (1′R,5R).13 This is one of only four monosubstituted BEDT-TTFs which have been prepared as a pure enantiomer.14 This BEDT-TTF is also novel in having a side-chain with an aromatic ring with potential to pack with planar acceptor molecules. We report here the first radical-cation salt of 1 which is a chiral conductor showing metallic behaviour down to 4.2 K. Most notably, this salt also undergoes an insulator to metal transition (IM) close to room temperature. Materials which are able to switch their properties depending on their temperature could lead to a new generation of electronic devices. Atom-by-atom changes to this material offers the potential for the synthesis of a family of materials each with a different IM transition temperature.
θ-(1)4TCNQ crystallises in a monoclinic crystal system in chiral space group C2 at 150 K, but in a triclinic system in space group P1 at 300 K with the a axis being halved.§ At 150 K the asymmetric unit consists of one donor molecule and a quarter of a TCNQ molecule, whilst at 300 K there is somewhat unusual disorder. In the donor stacking direction (along the b-axis) there are two donors with occupancies of 0.87 and 0.13, respectively. Both donors are too close to co-exist, since the b-axis length is only 4.8570(5) Å. The minor occupancy donor was fixed to have the same shape as that of the major donor. One possibility is that one donor stack is slipped along the b-axis compared to the other. Such disorder is commonly observed at room temperature where the occupancy of both donors is 0.5. Another possibility is the existence of a superstructure15 which will be discussed later.
 | ||
| Scheme 1 | ||
In both the 150 K and 300 K phases the thick anion sheet and the donor layer stack alternately along the a axis (Fig. 1) with the donors adopting an θ-type packing (Fig. 2). Several BEDT-TTF salts with θ-type packing are known, having monoclinic C2 space group at low temperature and orthorhombic I222 at room temperature. Mori et al.16 have previously shown a relationship between the dihedral angle between neighbouring donor stacks and the physical properties of the θ salts involving the series θ-(BEDT-TTF)2MM′(SCN)4 (M = Tl, Rb, Cs, M′ = Co, Zn). The ground state of these quarter-filled salts varies from insulating to metallic with decreasing dihedral angle. The metal–insulator transition temperature in this series of θ salts varies from 250 K down to 20 K as the dihedral angle changes from ∼116° to 105°. θ-(BEDT-TTF)2CsZn(SCN)4 has a dihedral angle of 105° and undergoes a metal–insulator transition at 20 K, whilst θ-(BEDT-TTF)2I3 has a dihedral angle of 99° and shows ambient pressure superconductivity at ∼4 K.17
 | ||
| Fig. 1 Structure of θ-(1)4TCNQ at 150 K viewed along the b axis showing the disordered phenyl groups and TCNQ molecules. | ||
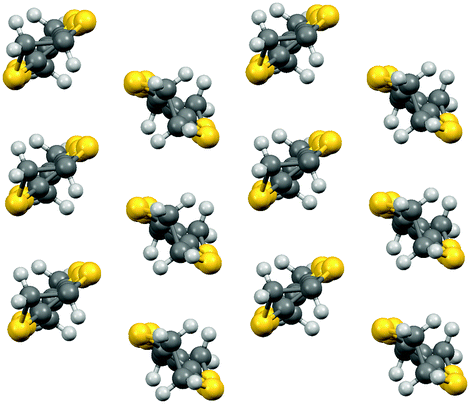 | ||
| Fig. 2 θ-type packing of donor molecules. | ||
SQUID magnetometry on θ-(1)4TCNQ showed no superconductivity down to 1.8 K. Four-probe DC transport measurements were made on four single crystals of θ-(1)4TCNQ and all showed metallic behaviour down to 4.2 K. However, in contrast with other θ salts we observe in θ-(1)4TCNQ both hysteresis and an insulator–metal transition (IM) at 285–290 K (Fig. 3). The following paragraphs discuss the structural features which lead to this IM transition.
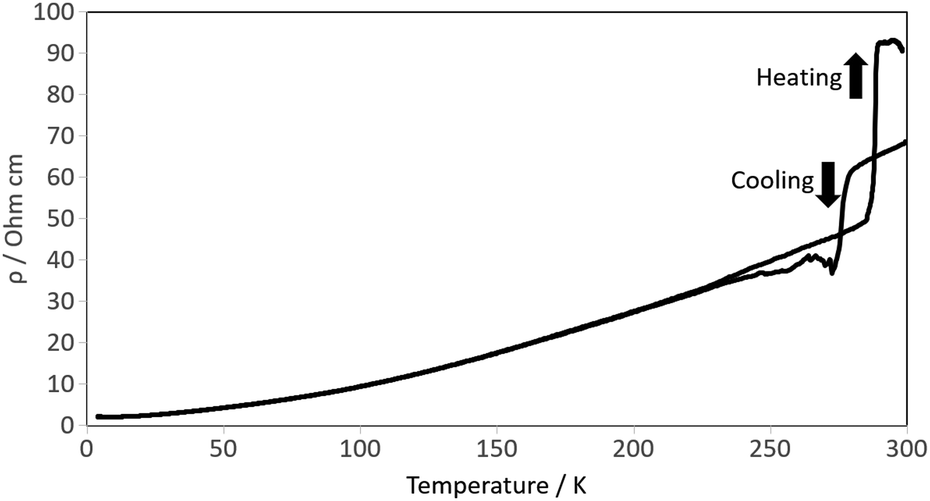 | ||
| Fig. 3 Electrical resistivity measured on a single crystal of θ-(1)4TCNQ. | ||
Raman spectroscopy of θ-(1)4TCNQ at 300 K shows only peaks corresponding to TCNQ−˙ (1387 cm−1 and 1613 cm−1), whilst cooling below the IM transition temperature leads to the appearance of additional peaks corresponding to TCNQ0 (1450 cm−1 and 1600 cm−1). This change in TCNQ charge is reversible upon repeated heating and cooling cycles. SQUID magnetometry also confirms the transfer of charge from TCNQ to donor below the IM transition temperature.
In the crystal structure solution at 150 K the –CH(CH3)phenyl group on the donor side chain appears disordered over two positions, with pairs of these side chains from different donor stacks butted up against each other within the TCNQ layer (Fig. 1). We interpret the disorder in the TCNQ stack along the c direction as the two different arrangements of these two neighbouring side chains, Ph1 and Ph2, alternating in the order –Ph1-Ph2-TCNQ-Ph1-Ph2-TCNQ– so only every other TCNQ is present (Fig. 4). The stacks along c above and below in the b direction, will be such that the TCNQs lie above the block of four side chains. The short b axis cannot accommodate adjacent TCNQs. This pattern is not in sync for all parallel layers, hence leading to the apparent disorder.
 | ||
| Fig. 4 Structure of θ-(1)4TCNQ at 150 K viewed along the b axis showing one of the orientations of the disordered phenyl groups and TCNQ molecule. | ||
The hysteresis of the resistivity curves indicates that the transition is a first-order transition. The salt is a stable metal from 250–4.2 K therefore at least from 150 to 2 K the donor layer should have a Pauli contribution to the magnetism which usually has no temperature dependence. Therefore, we fitted the magnetic data in a χT graph using a Curie–Weiss model with a temperature-independent factor a (χ = C/(T − θ) + a) leading to values of C = 0.228 emu K mol−1, θ = −0.09 K and a = 7.7 × 10−4 emu mol−1. The C value and the Raman results (TCNQ0 and TCNQ− co-exist) suggest that the average charge per TCNQ is found to be 0.61, which means approximately 6/10 TCNQ molecules are monoanionic and 4/10 are neutral in the lower temperature phase. The θ of almost zero indicates that the magnetic interactions between TCNQ monoanions are negligible, which suggests that the TCNQ0 and TCNQ− are almost alternately located in the TCNQ chain along the shortest b axis. This is to be expected because contacts between monoanions would provide a large Coulomb repulsion. It raises a question as to why the TCNQ chain does not select complete alternation, namely TCNQ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TCNQ− = 1:1 (the actual ratio is 2:3).
:TCNQ− = 1:1 (the actual ratio is 2:3).
The donor is chiral so all NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are directed along the b axis, the stacking direction, in the same sense – this means that the donor layer has a relatively large dipole moment ∼4 Debye, and voltage ∼0.5–0.6 eV. As a result, TCNQ chains should cancel the donor's dipole moment. We speculate that the TCNQ monoanions’ distribution is not equal but biased to cancel the dipole moments provided by the amide groups of the donor molecules, i.e. the NH side of the crystal includes more TCNQ− molecules than the opposite CO side of the crystal. Thus, the non-stoichiometric TCNQ charge is due to the donor chirality-induced dipole moment.
O groups are directed along the b axis, the stacking direction, in the same sense – this means that the donor layer has a relatively large dipole moment ∼4 Debye, and voltage ∼0.5–0.6 eV. As a result, TCNQ chains should cancel the donor's dipole moment. We speculate that the TCNQ monoanions’ distribution is not equal but biased to cancel the dipole moments provided by the amide groups of the donor molecules, i.e. the NH side of the crystal includes more TCNQ− molecules than the opposite CO side of the crystal. Thus, the non-stoichiometric TCNQ charge is due to the donor chirality-induced dipole moment.
The temperature-independent factor a is larger than that of a normal 2:1 salt (maybe double). However, the average donor charge of 0.15+ in this 4:1 salt is unusual implying that a large a value may be possible for such a unique situation. Next, we subtracted the a value from the total magnetic susceptibility to produce the χT–T plot in Fig. 5. This indicates that spin concentration decreases suddenly at about 270 K from 1.0 and becomes constant, ≈0.6. This strongly indicates that a charge-change (CC) transition occurs from TCNQ− only to the mixture of TCNQ0:TCNQ− = 1:1. Neutral-Ionic (NI) transitions (e.g. TTF-chloranil) are very well known but this is the first example of a CC transition to the best of our knowledge. This implies that at 300 K relatively large thermal motion cannot cancel the imbalance of the dipole moments between the donor and TCNQ chains as discussed in detail later.
 | ||
| Fig. 5 χT versus T for θ-(1)4TCNQ. | ||
Fig. S1 (ESI‡) shows the band calculations for θ-(1)4TCNQ at 300 K (top) and 150 K (bottom). The Fermi surfaces are simple at both temperatures indicating that the salt should be metallic. Owing to the incommensurate band filling of 0.92 at 150 K where TCNQ0.61−, it is impossible for them to become either band or Mott insulators. The 300 K case, which is in a commensurate insulating state, can be a Mott or a charge-ordered state, but it is somewhat unusual because all TCNQs are monoanions, whose negative charges are uniformly distributed in the crystal at room temperature. Indeed, a charge-ordered state should be observed at 150 K, where neutral and monoanionic TCNQ co-exist, which provide a periodic or random potential, but it is indeed a stable metallic phase.
The Ewald sphere of the room temperature phase is shown in Fig. S2 (ESI‡), which indicates the existence of extra spots at 300 K at h = 0.5n and k = 0.5n, where n = some odd numbers. This indicates the formation of donor octamers. The crystallographic data at 300 K indicates that the phenyl ring on the chiral carbon has very large temperature factors, suggesting that the phenyl rings rotate above the IM transition. This leads to weaker donor–donor interactions, i.e. this provides negative chemical pressure to the donor layer. The unit cell expansion might disturb the conduction pathway and the resistivity therefore increases. If this disorder is an eight-fold superstructure (Fig. S3, ESI‡), the octamer has 2 holes which could explain why the salt is an insulator above the IM transition temperature, which is confirmed by the extra reflections on the Ewald sphere. We therefore speculate that the IM transition is a phenyl-ring motion-induced insulator–metal (IM) transition. Normally an insulating state (I) has lower energy than a metallic state (M), therefore the transition from M to I is normal, and the I to M transition is quite unusual. However, the insulating state in this case is caused by the negative pressure due to cell expansion of the almost free rotation of phenyl rings, which has large entropy (≈R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln2). Therefore, the insulating state can exist at a higher temperature than that of the metallic state. Below the IM transition temperature a charge-change (CC) transition occurs and the chirality-induced polarity leads to the TCNQ chains adopting non-stoichiometric charge to create the opposite polarity, causing the donor layer to be metallic.
ln2). Therefore, the insulating state can exist at a higher temperature than that of the metallic state. Below the IM transition temperature a charge-change (CC) transition occurs and the chirality-induced polarity leads to the TCNQ chains adopting non-stoichiometric charge to create the opposite polarity, causing the donor layer to be metallic.
Bulk chiral molecular conductors are highly sought after because they can help formulate a theoretical model for eMChA. We report the synthesis of a chiral molecular metal which remains metallic down to at least 4.2 K. θ salts with BEDT-TTF0.5+ invariably lose their metallic state at some temperature, but the BEDT-TTF0.15+ salt reported here keeps the metallic state down to very low temperatures. This opens the possibility of controlling the band filling as a way to reach superconductivity in θ salts. We are currently synthesising structurally related donor molecules, including the other three stereoisomers of this salt, and investigating other methods for synthesis of salts of this type.
This salt shows the first example of a charge-change (CC) transition to the best of our knowledge. Below the insulator–metal transition temperature the chirality-induced donor amide polarity leads to the TCNQ chains adopting non-stoichiometric charge to create the opposite polarity, causing the donor layer to be metallic.
This salt also exhibits room-temperature switching capabilities with a transition from the insulating to the metallic state upon cooling below 10 °C. Materials which are able to switch their properties depending on their temperature could lead to a new generation of electronic devices. Atom-by-atom changes to this material offers the potential for the synthesis of a family of materials each with a different IM transition temperature.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Qin, W. Shi, T. Ideue, M. Yoshida, A. Zak, R. Tenne, T. Kikitsu, D. Inoue, D. Hashizume and Y. Iwasa, Nat. Commun., 2017, 8, 1–6 CrossRef PubMed.
- G. L. J. A. Rikken, J. Fölling and P. Wyder, Phys. Rev. Lett., 2001, 87, 236602 CrossRef CAS PubMed; G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493–494 CrossRef.
- V. Krstic, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315–11319 CrossRef CAS; V. Krstić and G. L. J. A. Rikken, Chem. Phys. Lett., 2002, 364, 51–56 CrossRef; G. L. J. A. Rikken, Science, 2011, 331, 864–865 CrossRef PubMed.
- M. Atzori, F. Pop, P. Auban-Senzier, C. J. Gómez-García, E. Canadell, F. Artizzu, A. Serpe, P. Deplano, N. Avarvari and M. L. Mercuri, Inorg. Chem., 2014, 53, 7028–7039 CrossRef CAS PubMed; M. Atzori, F. Pop, P. Auban-Senzier, R. Clérac, E. Canadell, M. L. Mercuri and N. Avarvari, Inorg. Chem., 2015, 54, 3643–3653 CrossRef PubMed; J. R. Lopez, L. Martin, J. D. Wallis, H. Akutsu, Y. Nakazawa, J. I. Yamada, T. Kadoya, S. J. Coles and C. Wilson, Dalton Trans., 2016, 45, 9285–9293 RSC.
- L. Martin, Coord. Chem. Rev., 2018, 376, 277–291 Search PubMed.
- L. Martin, P. Day, H. Akutsu, J. I. Yamada, S. Nakatsuji, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, P. McMillan and S. Firth, CrystEngComm, 2007, 9, 865–867 RSC.
- L. Martin, H. Akutsu, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2015, 17, 2783–2790 RSC; L. Martin, H. Akutsu, P. N. Horton, M. B. Hursthouse, R. W. Harrington and W. Clegg, Eur. J. Inorg. Chem., 2015, 1865–1870 CrossRef CAS.
- N. Mroweh, F. Pop, C. Mézière, M. Allain, P. Auban-Senzier, N. Vanthuyne, P. Alemany, E. Canadell and N. Avarvari, Cryst. Growth Des., 2020, 20, 2516–2526 CrossRef CAS; N. Mroweh, P. Auban-Senzier, N. Vanthuyne, E. Canadell and N. Avarvari, J. Mater. Chem. C, 2019, 7, 12664–12673 RSC; F. Pop, P. Auban-Senzier, E. Canadell and N. Avarvari, Chem. Commun., 2016, 52, 12438–12441 RSC.
- S. Yang, F. Pop, C. Melan, A. C. Brooks, L. Martin, P. Horton, P. Auban-Senzier, G. L. J. A. Rikken, N. Avarvari and J. D. Wallis, CrystEngComm, 2014, 16, 3906–3916 RSC; S. Yang, A. C. Brooks, L. Martin, P. Day, M. Pilkington, W. Clegg, R. W. Harrington, L. Russo and J. D. Wallis, Tetrahedron, 2010, 66, 6977–6989 CrossRef CAS; R. J. Brown, A. C. Brooks, J. P. Griffiths, B. Vital, P. Day and J. D. Wallis, Org. Biomol. Chem., 2007, 5, 3172–3182 RSC.
- J. R. Galáln-Mascarós, E. Coronado, P. A. Goddard, J. Singleton, A. I. Coldea, J. D. Wallis, S. J. Coles and A. Alberolá, J. Am. Chem. Soc., 2010, 132, 9271–9273 CrossRef CAS PubMed; F. Pop, S. Laroussi, T. Cauchy, C. J. Gomez-Garcia, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466–474 CrossRef PubMed.
- F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 1–6 CAS.
- F. Pop, P. Auban-Senzier, A. Fŗckowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed.
- I. Awheda, S. J. Krivickas, S. Yang, L. Martin, M. A. Guziak, A. C. Brooks, F. Pelletier, M. Le Kerneau, P. Day, P. N. Horton, H. Akutsu and J. D. Wallis, Tetrahedron, 2013, 69, 8738–8750 CrossRef CAS.
- Q. Wang, P. Day, J. P. Griffiths, H. Nie and J. D. Wallis, New J. Chem., 2006, 30, 1790–1800 RSC; J. P. Griffiths, H. Nie, R. J. Brown, P. Day and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 2155–2166 RSC; T. Ozturk, C. R. Rice and J. D. Wallis, J. Mater. Chem., 1995, 5, 1553–1556 RSC.
- A. Kobayashi, T. Naito and H. Kobayashi, Synth. Met., 1995, 70, 1047–1048 CrossRef.
- T. Mori, H. Mori and S. Tanaka, Bull. Chem. Soc. Jpn., 1999, 72, 179–197 CrossRef; H. Mori, S. Tanaka and T. Mori, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 12023–12029 CrossRef; R. Kondo, M. Higa, S. Kagoshima, H. Hoshino, T. Mori and H. Mori, J. Phys. Soc. Jpn. DOI:10.1143/JPSJ.75.044716.
- H. Kobayashi, R. Kato, A. Kobayashi, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1986, 789–792 CrossRef CAS; H. Kobayashi, R. Kato, A. Kobayashi, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1986, 833–836 CrossRef; R. Kato, H. Kobayashi, A. Kobayashi, Y. Nishio, K. Kajita and W. Sasaki, Chem. Lett., 1986, 957–960 CrossRef; K. Kajita, Y. Nishio, S. Moriyama, W. Sasaki, R. Kato, H. Kobayashi and A. Kobayashi, Solid State Commun., 1987, 64, 1279–1284 CrossRef.
Footnotes |
| † This article is dedicated to Professor Peter Day. LM and TB would like to thank the Royal Society and Leverhulme Trust for financial support (LT170022 and RPG-2019-242). |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S3. CCDC 1984873 and 1984874. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04094k |
| § 45.7 mg of donor 1 and 17.6 mg TCNQ in 13 ml dichloromethane was heated under reflux for two hours whilst stirring. Slow evaporation gave black starbursts of crystalline needles of a 4:1 salt of donor 1 with TCNQ. Crystal data at 150 K: (C20H19S8N1O1)4·C12H4N4, M = 2387.62, black needle, a = 46.8847(19) Å, b = 4.9361(2) Å, c = 11.2909(5) Å, β = 99.121(7)°, U = 2580.0(2) Å3, T = 150 K, space group C2, Z = 1, μ = 7.136 mm−1, reflections collected = 12105, independent reflections = 5838, R1 = 0.0442, wR2 = 0.1289 [F2 > 2σ(F2)], R1 = 0.0515, wR2 = 0.1323 (all data). CCDC 1984873.‡ Crystal data at 300 K: (C20H19S8N1O1)2·0.5(C12H4N4), M = 1193.81, black needle, a = 23.723(2) Å, b = 4.8570(5) Å, c = 11.6968(10) Å, α = 89.841(6)°, β = 81.637(6)°, γ = 87.168(6)°, U = 1331.8(2) Å3, T = 300 K, space group P1, Z = 1, μ = 6.912 mm−1, reflections collected = 12845, independent reflections = 9915, R1 = 0.0997, wR2 = 0.2713 [F2 > 2σ(F2)], R1 = 0.1474, wR2 = 0.3145 (all data). CCDC 1984874.‡ |
| This journal is © The Royal Society of Chemistry 2020 |