
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic access to a phosphorescent non-palindromic pincer complex of palladium by a double oxidative addition – comproportionation sequence†
Wolfram
Feuerstein
 and
Frank
Breher
*
and
Frank
Breher
*
Karlsruhe Institute of Technology (KIT), Institute of Inorganic Chemistry, Division Molecular Chemistry, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.edu
First published on 26th August 2020
Abstract
A highly luminescent non-palindromic [(C^C^N)Pd] pincer complex forms upon reacting pyridine-substituted 2,2′-diiodo-biphenyl with [Pd(PPh3)4]. This case study establishes for the first time that the title compound is formed via a double oxidative addition – comproportionation sequence. DFT and TDDFT calculations complement mechanistic and photophysical characterizations.
Pincer complexes1 of platinum and gold gained increasing attention during the past 30 years due to their simple preparation2 and appealing photophysical properties making them interesting for applications in photocatalysis3 and electroluminescent devices,4,5 but in the field of catalysis6 and biomedical applications7 as well. Contrasting the plethora of investigations concerned with diphenylpyridine-based (C^N^C) pincer complexes, only one example of palladium (1) has been published in the literature until now (Fig. 1).8 Missing investigations on the (C^N^C) palladium complexes are, among other, mainly caused by the lack of feasible methods for their synthesis reported so far, and the fact that the only photophysically investigated complex [(C^N^C)PdII(py)] (py = pyridine) exhibits only poor luminescence properties in contrast to its platinum analogue.9 Nevertheless, there are few reports on other emissive Pd complexes.10 Che and co-workers developed tetradentate (C^C^N^O) ligands and applied them for the synthesis of PdII complexes highly phosphorescent in solution at room temperature.11 A tetradentate ligand scaffold is certainly superior to tridentate pincers regarding the ability to suppress vibrational relaxation pathways of photoexcited states due to the even more rigid nature. However, tetradentate ligands occupy all coordination sites of a square-planar complex, which may not be desirable in view of potential catalytic applications.12 For example, the (C^N^C) moiety was proven to show very promising chemical properties at ligated AuIII centres making them interesting for new chemical transformations.13
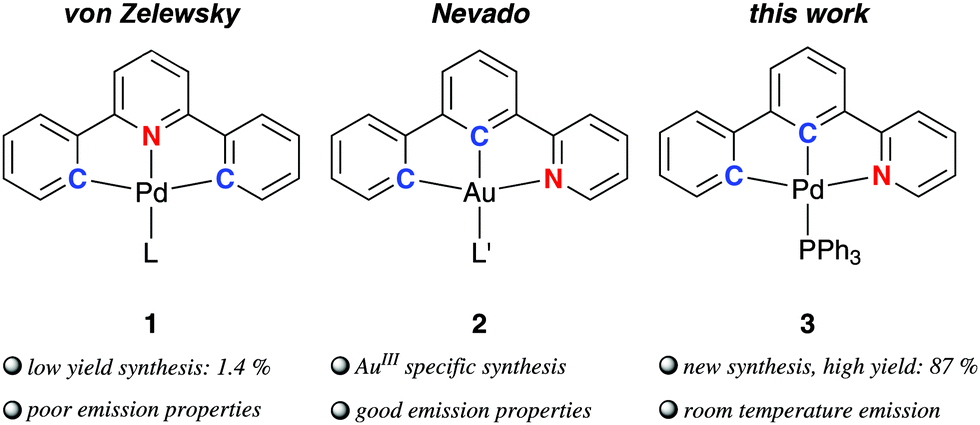 | ||
| Fig. 1 [(C^N^C)PdII] pincer complex 1 (L = SEt2, pyridine) and [(C^C^N)AuIII] complexes 2 (L′ = Cl, F, acetylide, aryl) reported in the literature; complex 3 described in this study. | ||
In 2015, Nevado and co-workers described a non-palindromic12,14 biphenyl-based (C^C^N) ligand employed for cyclometalated AuIII complexes (2 in Fig. 1).15 The non-palindromic (C^C^N) pincer ligand differs from the palindromic (C^N^C) motif regarding chemical properties due to the stronger trans influence of the carbon donor of the central phenyl ring.15,16 In addition, the [(C^C^N)AuIII] complexes outperform the majority of [(C^N^C)AuIII] analogues regarding emission quantum yields making them attractive for optoelectronic devices.17 The higher emission efficiency was traced back to stronger ligand field splitting of the non-palindromic (C^C^N) scaffold compared to the (C^N^C) analogue (cf. Section S5, ESI† for an in-depth discussion).15,18
Here, we present a simple procedure for the preparation of a non-palindromic [(C^C^N)PdII] complex based on a double oxidative addition of 2,2′-diiodobiphenyl substituted in 3-position with pyridine. This simple high-yield synthesis of a palladium(II) pincer complex 3 employing two phenyl rings and one pyridine donor makes this structural motif accessible for investigations and applications, which are until now mainly covered by platinum, gold and other transition metals.19 We use the analysis of photophysical properties as tool to shed light on the non-palindromic nature and impact of the (C^C^N) motif.
In a recent study, we have described the synthesis of pyridine-substituted 2,2′-diiodobiphenyl 5 as suitable pre-ligand for the preparation of [(C^C^N)AuIII] complexes.20 Our initial intention was to use this diiodobiphenyl for the preparation of a PdIV complex by double oxidative addition of Pd0 into both iodophenyl groups of 5. Bautista and co-workers reported on the oxidative addition of aryl iodides to PdII to readily happen if the aryl iodide exhibits a functional group, which is able to coordinate to the PdII centre.21 Thus, we reacted 5 with [Pd(PPh3)4] in toluene (Scheme 1).
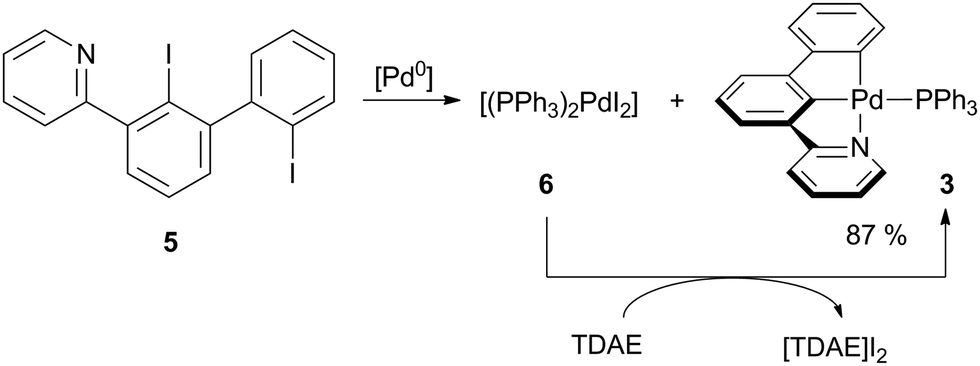 | ||
| Scheme 1 Synthesis of [(C^C^N)PdII] complex 3. [Pd0] = [Pd(PPh3)4]. Conditions: toluene, 100 °C, 4 h. | ||
Upon heating, the formerly yellow solution turned dark red (Fig. S1, ESI†). After workup, two species could be isolated in equal amounts by manually picking crystals: [(PPh3)2PdI2] (6) and the palladium(II) pincer complex 3.
Full conversion of pre-ligand 5 was achieved by addition of one equivalent of tetrakis(dimethylamino)ethylene (TDAE) to reduce 6in situ.22 Following this procedure, the pincer complex 3 was obtained in 87% overall yield in form of a yellow microcrystalline solid, which is luminescent under UV irradiation. Instead of [Pd(PPh3)4], the dichloride [(PPh3)2PdCl2] may also be used when employing two equivalents TDAE.
The structure of 3 in the solid-state (Fig. 2) shows the palladium centre being coordinated in a distorted square-planar fashion. The long Pd–P bond length of 236.8 pm is indicative for the strong trans influence of the carbon donor of the central phenyl ring and is comparable to the Pd–P bond length of typical Pd0 phosphine complexes.23 The calculated (RI-TPSS-D3(BJ)/def2-TZVP) Pd–P bond length of the hypothetical palindromic analogue [(C^N^C)PdIIPPh3] was found to be 224 pm, i.e., 12 pm shorter than experimentally found for 3 (calculated: 236 pm).
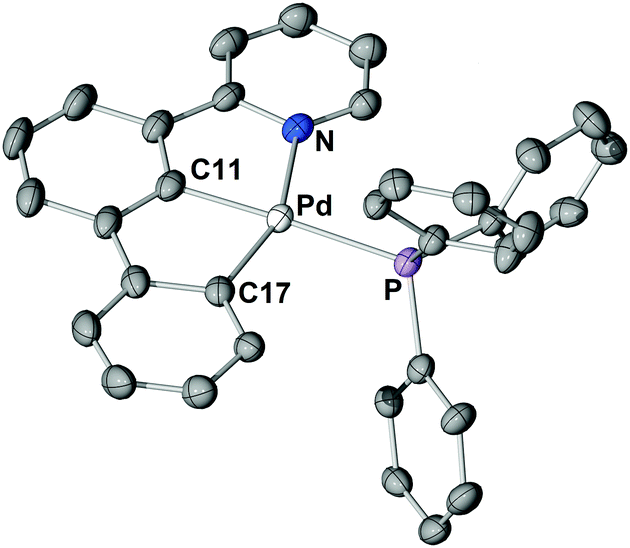 | ||
| Fig. 2 Molecular structure of 3. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Only one molecule of the asymmetric unit is shown. Selected bond lengths (pm) and angles (°): Pd–P 236.8(9), Pd–N 218.8(3), Pd–C11 197.3(4), Pd–C17 204.0(4), N–Pd–C11 78.3(2), C11–Pd–C17 79.7(2). | ||
The simultaneous formation of pincer complex 3 in oxidation state +II and the diiodide 6a priori suggested the intermediary formation of a PdIV species and their comproportionation with not yet reacted Pd0 precursors. In principle, a bimetallic process involving the oxidative addition of two different metal centres into the two iodophenyl groups of 5 and subsequent intramolecular transmetalation could also be plausible. Such a bimetallic process should lead to a mixture of non-, mono- and diactivated 5 and derived products. However, the reaction compiled in Scheme 1 always yielded equal amounts of 3 and diiodide 6 with approximately 50% conversion of pre-ligand 5, which disagrees with a bimetallic mechanism.
To shed further light on this reaction, we performed a control experiment and reacted 2,2′-diiodobiphenyl (S1) with [Pd(PPh3)4] (Scheme S2, ESI†) upon which the diiodide 6 formed as well. However, the other product(s) of this reaction, a yellow solid, we were not able to identify. 31P NMR spectroscopic investigations showed more than ten different NMR signals. Thus, the lateral pyridine donor of 5 is crucial for the formation of 3.
We further investigated the mechanistic pathway in more detail by DFT calculations (Scheme 2). Additional pathways are discussed in the Supporting Information (Section S6, ESI†).
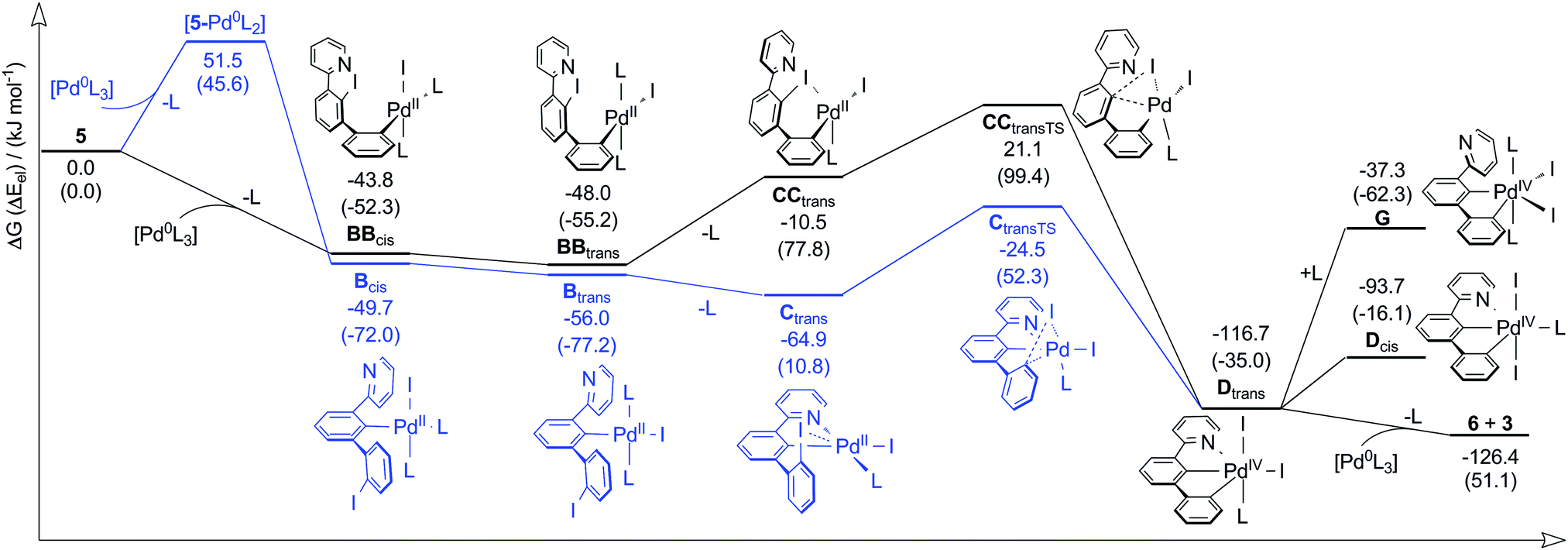 | ||
| Scheme 2 Mechanism of formation of 3 by comproportionation of intermediate PdIV species Dtrans and Pd0. The pathway starting by oxidative addition of the central aryl iodide is coloured in blue. All calculations were done on the RI-TPSS-D3(BJ)/def2-TZVP (SMD: toluene) level of theory at 298.15 K and 0.1 MPa. L = PPh3. ΔG: Gibbs free energy. Electronic energies Eel are given in brackets. Additional pathways of higher energy intermediates and computational details are given in the Supporting Information. | ||
[Pd(PPh3)4] is known to lose one or two ligands upon solvation making the Pd0 atom reactive for oxidative addition.24 We assume that, in a first step, [Pd0(PPh3)3] oxidatively adds into the C–I bond of the central (Bcis) or lateral (BBcis) iodophenyl group of 5 with loss of one PPh3 ligand (L). A coordination of Pd0 to the pyridine donor ([5-PdL3]) does probably not precede a central addition since the hard donor pyridine will most likely not replace the softer PPh3. Interestingly, the formation of Bcis is slightly favoured over BBcis although the former is more sterically hindered. However, we identified dispersion interactions responsible for the opposite finding since optimization of structures without dispersion-corrected DFT25 predicts the lateral addition to be favoured by −10.9 kJ mol−1. Dispersion interaction was shown to often be responsible for chemical properties standing in contrast to considerations only based on a molecule's bulk, e.g. the dimerization of the (bulky) tert-butyl substituted Gomberg radical.26
After a first oxidative addition‡ and cis–trans isomerization (Btrans/BBtrans) one PPh3 is displaced leading to the PdII species Ctrans and CCtrans, respectively. The former is highly stabilized due to (C^N) chelation. Thus, CCtrans is calculated to be 54.4 kJ mol−1 higher in energy than Ctrans. We note that (C^N) cyclometalation of Bcis may precede cis–trans isomerization (cf. Scheme S5, ESI†). Species Ctrans and CCtrans undergo a second oxidative addition to form the same PdIV intermediate Dtrans, stabilized by the (C^C^N) pincer. A species G with a second coordinated PPh3 instead of pyridine is disfavoured by an energy of 79.4 kJ mol−1. Thus, the pyridine donor not only lowers the energy of intermediates of the pathway involving central oxidative addition (blue), but also stabilizes PdIV intermediates as well. Finally, Dtrans readily comproportionates with [Pd(PPh3)3] to form the final products 6 and 3.
Pincer complex 3 shows a blue shifted absorption in the less polar solvent CH2Cl2 compared to tetrahydrofurane (THF) indicating a charge-transfer character of the first absorption bands (Fig. 3). Slight vibronic structures with line spacings of ca. 1300 cm−1, i.e., the frequency of pincer breathing modes,5,27 indicate π → π* IL/MLCT transitions. This assignment is supported by TD-DFT calculations (Section S4, ESI†).
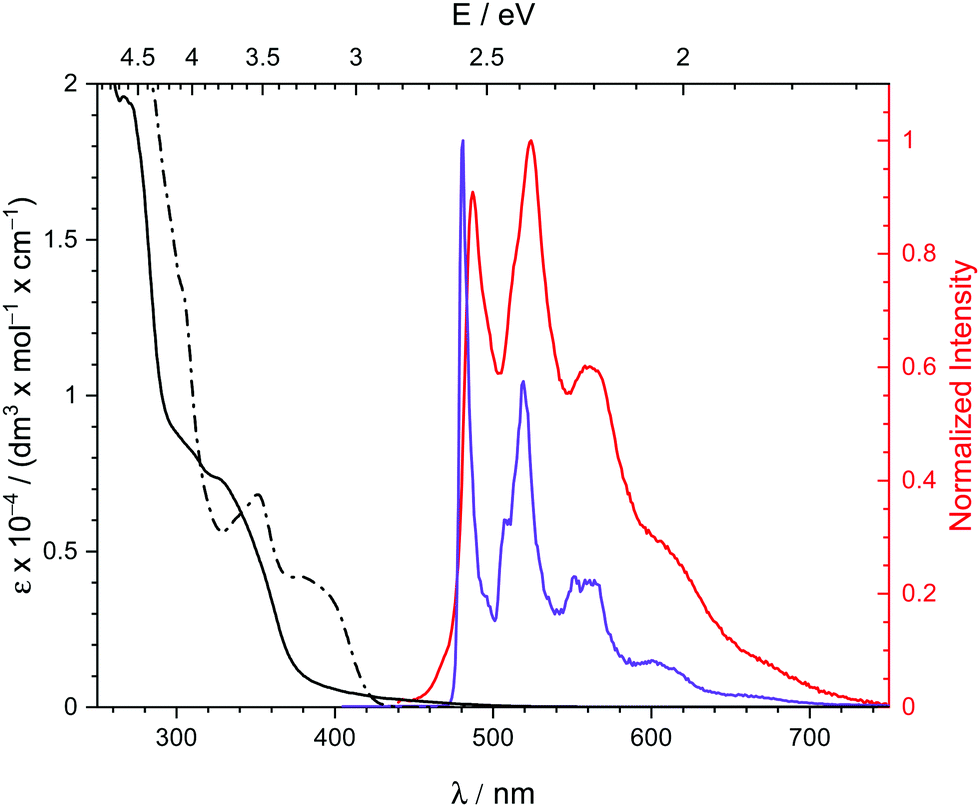 | ||
| Fig. 3 Absorption spectra of [(C^C^N)Pd(PPh3)] (3) in CH2Cl2 (straight line) and THF (dotted line) and photoemission spectrum in the solid state at 293 K (red) and in frozen THF solution at 77 K (purple). Spectra were recorded under oxygen-free conditions. | ||
Complex 3 exhibits intense yellow phosphorescence in the solid-state at room temperature with a quantum yield of 10%. This finding underlines the extraordinary electronic impact of the non-palindromic pincer since palladium complexes luminescent at room temperature are rare.10 An emission lifetime of 0.4 μs is indicative for a phosphorescence process. 3 shows no luminescence in liquid solution, probably due to structural distortions in the excited state.28 However, in frozen THF solution at 77 K we observe similar emission with a notably longer lifetime of 370 μs, thus, standing in line with a decreased vibronic relaxation. The vibronic structured emission of 3 (Fig. 3, red and purple) is comparable to the signature of other (C^C) and (C^N^C) cyclometalated complexes and indicates a metal-perturbed, pincer-centred π* → π transition.5,27 Again, this is supported by TD-DFT calculations (Fig. 4) showing the donor orbital of the S0 ← T1 transition being a π* orbital of the pincer with major contribution of the pyridine ring.
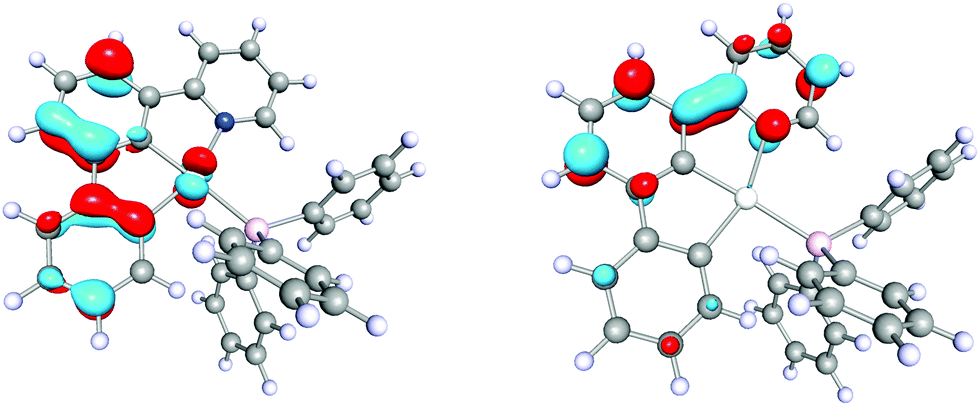 | ||
| Fig. 4 Spin orbitals (isovalue: 0.06 a.u.) of the phosphorescence transition S0 ← T1 of [(C^C^N)Pd(PPh3)] (3) calculated by Spin–Flip-TDDFT (RI-PWLDA/def2-TZVP). Orbital contribution (square of orbital coefficients): 99.6%. Calculated S0 ← T1 transition energy: −2.42 eV (T1 optimized structure). Left: 152β (acceptor orbital), right: 153α (donor orbital). | ||
In summary, a non-palindromic (C^C^N) complex of palladium with outstanding luminescence properties can be prepared by means of a double oxidative addition – comproportionation sequence of 3-pyridine-substituted 2,2′-diiodobiphenyl 5 and [Pd(PPh3)4]. The presented approach complements already known procedures for the synthesis of pincer complexes and extends the field of pincers invoking two phenyl donors to palladium. We could illustrate the strong trans influence of the carbon donor of the central phenyl ring of the pincer ligand, which may point to unique catalytic16 and biological29 properties currently under investigation in our group.
In the future, the presented synthetic principle may prove its viability in the development of pincer complexes invoking other metals than palladium as well; first promising results for nickel are shown in the Supplementary Information (Section S3, ESI†).
W. F. thanks the Carl-Zeiss Stiftung for a PhD scholarship and the Studienstiftung des Deutschen Volkes for general support. We thank Dr Lilli Neumeier for determination of emission quantum yields and Dr Sergei Lebedkin for the recording of emission spectra at low temperature.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. van Koten, J. Organomet. Chem., 2013, 730, 156 Search PubMed; G. van Koten, Pure Appl. Chem., 1989, 61, 1681 Search PubMed.
- G. W. V. Cave, N. W. Alcock and J. P. Rourke, Organometallics, 1999, 18, 1801 Search PubMed; V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 117, 3167 Search PubMed.
- A. M. Ranieri, L. K. Burt, S. Stagni, S. Zacchini, B. W. Skelton, M. I. Ogden, A. C. Bissember and M. Massi, Organometallics, 2019, 38, 1108 Search PubMed; Z. Li, Y. Han, Z. Gao and F. Wang, ACS Catal., 2017, 7, 4676 Search PubMed.
- M. C. Tang, A. K. Chan, M. Y. Chan and V. W. Yam, Top. Curr. Chem., 2016, 374, 46 Search PubMed; M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 Search PubMed; M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115 Search PubMed; E. Turner, N. Bakken and J. Li, Inorg. Chem., 2013, 52, 7344 Search PubMed; C. Cebrian and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459 Search PubMed; C. Lee, R. Zaen, K.-M. Park, K. H. Lee, J. Y. Lee and Y. Kang, Organometallics, 2018, 37, 4639 Search PubMed.
- S. C. Kui, F. F. Hung, S. L. Lai, M. Y. Yuen, C. C. Kwok, K. H. Low, S. S. Chui and C. M. Che, Chem. – Eur. J., 2012, 18, 96 Search PubMed.
- M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 Search PubMed; P. A. Shaw, G. J. Clarkson and J. P. Rourke, Chem. Sci., 2017, 8, 5547 Search PubMed; P. Hamidizadeh, S. M. Nabavizadeh and S. J. Hoseini, Dalton Trans., 2019, 48, 3422 Search PubMed; D. Serra, P. Cao, J. Cabrera, R. Padilla, F. Rominger and M. Limbach, Organometallics, 2011, 30, 1885 Search PubMed.
- S. Jurgens, F. E. Kuhn and A. Casini, Curr. Med. Chem., 2018, 25, 437 Search PubMed; S. Garbe, M. Krause, A. Klimpel, I. Neundorf, P. Lippmann, I. Ott, D. Brünink, C. A. Strassert, N. L. Doltsinis and A. Klein, Organometallics, 2020, 39, 746 Search PubMed; N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodríguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784 Search PubMed; C. K. Li, R. W. Sun, S. C. Kui, N. Zhu and C. M. Che, Chem. – Eur. J., 2006, 12, 5253 Search PubMed; H. Luo, B. Cao, A. S. C. Chan, R. W. Y. Sun and T. Zou, Angew. Chem., Int. Ed., 2020, 132, 11139 Search PubMed.
- C. Cornioley-Deuschel, T. Ward and A. Von Zelewsky, Helv. Chim. Acta, 1988, 71, 130 Search PubMed.
- M. Maestri, C. Deuschel-Cornioley and A. Von Zelewsky, Coord. Chem. Rev., 1991, 111, 117 Search PubMed.
- M. Ghedini, D. Pucci, G. Calogero and F. Barigelletti, Chem. Phys. Lett., 1997, 267, 341 Search PubMed; F. Neve, A. Crispini, C. Di Pietro and S. Campagna, Organometallics, 2002, 21, 3511 Search PubMed; P. K. Chow, W. P. To, K. H. Low and C. M. Che, Chem. – Asian J., 2014, 9, 534 Search PubMed.
- P. K. Chow, C. Ma, W. P. To, G. S. Tong, S. L. Lai, S. C. Kui, W. M. Kwok and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775 Search PubMed; P. K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W. M. Kwok, W. H. Ang, C. Y. Chung, C. Yang, F. Wang and C. M. Che, Chem. Sci., 2016, 7, 6083 Search PubMed.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959 Search PubMed.
- D. A. Rosca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247 Search PubMed; D. A. Rosca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 Search PubMed; L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobarik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287 Search PubMed; N. Savjani, D. A. Rosca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 Search PubMed; L. Rocchigiani, J. Fernandez-Cestau, G. Agonigi, I. Chambrier, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2017, 56, 13861 Search PubMed.
- W. K. Kwok, M. C. Tang, S. L. Lai, W. L. Cheung, L. K. Li, M. Ng, M. Y. Chan and V. W. Yam, Angew. Chem., Int. Ed., 2020, 59, 9684 Search PubMed; L.-K. Li, M.-C. Tang, S.-L. Lai, M. Ng, W.-K. Kwok, M.-Y. Chan and V. W.-W. Yam, Nat. Photonics, 2019, 13, 185 Search PubMed.
- R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287 Search PubMed.
- H. Beucher, E. Merino, A. Genoux, T. Fox and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 9064 Search PubMed.
- H. Beucher, S. Kumar, E. Merino, W.-H. Hu, G. Stemmler, S. Cuesta-Galisteo, J. A. González, J. Jagielski, C.-J. Shih and C. Nevado, Chem. Mater., 2020, 32, 1605 Search PubMed.
- G. S. Ming Tong, K. T. Chan, X. Chang and C. M. Che, Chem. Sci., 2015, 6, 3026 Search PubMed; B. Z. Yang, X. Zhou, T. Liu, F. Q. Bai and H. X. Zhang, J. Phys. Chem. A, 2009, 113, 9396 Search PubMed; E. S. Lam, W. H. Lam and V. W. Yam, Inorg. Chem., 2015, 54, 3624 Search PubMed.
- J. A. Williams, Chem. Soc. Rev., 2009, 38, 1783 Search PubMed; S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 Search PubMed; M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970 Search PubMed; C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 Search PubMed; M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898 Search PubMed.
- W. Feuerstein, C. Holzer, X. Gui, L. Neumeier, W. Klopper and F. Breher, Chem. – Eur. J., 2020 DOI:10.1002/chem.202003271.
- J. Vicente, A. Arcas, F. Julia-Hernandez and D. Bautista, Angew. Chem., Int. Ed., 2011, 50, 6896 Search PubMed; I. Vicente-Hernandez, M. T. Chicote, J. Vicente and D. Bautista, Chem. Commun., 2016, 52, 594 Search PubMed.
- J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384 Search PubMed; N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 Search PubMed.
- V. S. Sergienko and M. A. Porai-Koshits, J. Struct. Chem., 1988, 28, 548 Search PubMed.
- J. A. Labinger, Organometallics, 2015, 34, 4784 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed; S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 Search PubMed.
- S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639 Search PubMed; S. Rosel, C. Balestrieri and P. R. Schreiner, Chem. Sci., 2017, 8, 405 Search PubMed.
- S. Fuertes, S. K. Brayshaw, P. R. Raithby, S. Schiffers and M. R. Warren, Organometallics, 2011, 31, 105 Search PubMed; R. Wai-Yin Sun, A. Lok-Fung Chow, X.-H. Li, J. J. Yan, S. Sin-Yin Chui and C.-M. Che, Chem. Sci., 2011, 2, 728 Search PubMed; B. Fang, Y. Zhu, L. Hu, Y. Shen, G. Jiang, Q. Zhang, X. Tian, S. Li, H. Zhou, J. Wu and Y. Tian, Inorg. Chem., 2018, 57, 14134 Search PubMed.
- C. J. Ballhausen, N. Bjerrum, R. Dingle, K. Eriks and C. R. Hare, Inorg. Chem., 1965, 4, 514 Search PubMed; C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409 Search PubMed.
- X. Riera, V. Moreno, C. J. Ciudad, V. Noe, M. Font-Bardia and X. Solans, Bioinorg. Chem. Appl., 2007, 2007, 98732 Search PubMed.
- M. Busch, M. D. Wodrich and C. Corminboeuf, ACS Catal., 2017, 7, 5643 Search PubMed; J. K. Stille and K. S. Y. Lau, Acc. Chem. Res., 2002, 10, 434 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR data, X-ray crystal structure data, quantum chemical calculations. CCDC 1986487 and 1986490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04065g |
| ‡ We have not determined the transition state of the first oxidative addition, since the oxidative addition of aryl iodides to Pd0 is known to readily happen.30 |
| This journal is © The Royal Society of Chemistry 2020 |