
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precise tetrafunctional streptavidin bioconjugates towards multifaceted drug delivery systems†
Dongdong
Xu
a,
Astrid Johanna
Heck
a,
Seah Ling
Kuan
 ab,
Tanja
Weil
ab and
Seraphine V.
Wegner
*ac
ab,
Tanja
Weil
ab and
Seraphine V.
Wegner
*ac
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
bInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cInstitute of Physiological Chemistry and Pathobiochemistry, University of Münster, Waldeyerstr. 15, 48149 Münster, Germany. E-mail: wegnerse@exchange.wwu.de
First published on 16th July 2020
Abstract
The preparation of precise macromolecules with multiple functionalities remains a challenge in drug delivery. Here, a method to prepare stoichiometrically precise tetrafunctional streptavidin conjugates is presented with an exemplary structure combining exactly one fluorescent label, one cell targeting group, one nucleus penetrating peptide and one drug molecule.
In drug delivery, the availability of macromolecules combining several different functionalities is often crucial to impart bioactivity, traceability and labelling. Beside the cytotoxicity of a drug designed as an anti-cancer agent, its targeted delivery to the right cell type and intracellular location as well as the real-time monitoring of the therapeutic are all desired features to increase the efficacy and safety.1 Yet, combining multiple functional groups in a single macromolecule is non-trivial.2–6 Multifunctionalization has mostly been realized with nanocarriers7 that are large enough to harbor many molecules but with limited control of the number of functional groups that are incorporated. These have been decorated with targeting ligands like antibodies8 or peptides9 and loaded with known small molecule drugs and imaging probes.10,11 Macromolecular drugs with many functionalities are far less common, due to the challenges in synthesis, solubility and preserving the activity. Even more important, general methods for the straightforward optimization of multifunctional molecules that would allow for efficient design and screening are missing.
Multivalent protein assemblies, which provide the advantages of simple production, monodisperse size and structure,12 have been developed for specific targeting and delivery. In particular, streptavidin with its four independent biotin binding pockets, extraordinarily high affinity (Kd = 10−14) and low exchange rates is an attractive scaffold.13 Moreover, the mature biotin labelling technology provides a large repertoire of biotinylated molecules with targeting, sensing, diagnostic and therapeutic entities. Yet, it is still challenging to produce structurally defined streptavidin conjugates with more than one type of biotin labeled molecule and precise stoichiometry. More precisely, when four different biotinylated molecules are mixed with streptavidin, a statistical mixture forms where only 1 of the 35 possible products is the desired tetrafunctional one. It is challenging if not impossible to separate the desired product, which contains all four functional labels from the mixture of defective structures and would result in extremely low yield. So far, only precise streptavidin conjugates with four different single stranded DNA have been prepared, which limits the type of functional groups in the macromolecular assembly.6 Other approaches to deal with multivalency or desymmetrization of streptavidin include the development of monovalent streptavidin to specifically label one receptor at the cell surface without artificially inducing clustering14 and to use solid support-based assemblies to sterically block some of the biotin binding sites.15 Nevertheless, incorporating more than two functional entities without restriction on one streptavidin in a controlled way is still elusive.
Herein, we describe a general method to prepare stoichiometrically exact tetrafunctional streptavidin conjugates offering great flexibility in conjugated molecules (Fig. 1A). For example, we combined a fluorescent label (atto-565-biotin), a cell type specific targeting group (folic acid–biotin), a nuclear localization peptide (nucleus penetrating peptide–biotin)16 and an anti-cancer drug (doxorubicin–biotin) within one bioconjugate, which allowed for specific and efficient cellular toxicity towards cancer cells and its simultaneous detection.
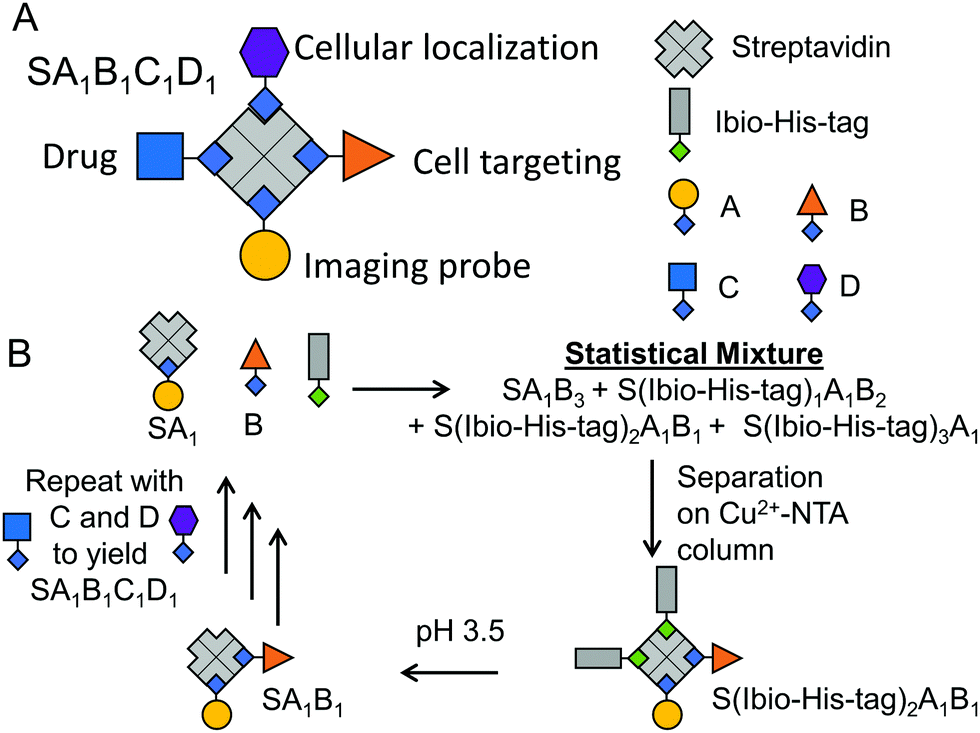 | ||
| Fig. 1 Strategy for preparing tetrafunctional streptavidin–biotin conjugates. (A) Four different biotinylated molecules for therapeutic activity, cell targeting, intracellular localization and theranostics are combined into one streptavidin conjugate. (B) Each of the functionalities is added one by one going through cycles of forming statistical mixtures of products containing different numbers of Ibio-His-tags, which are separated on a Cu2+-NTA column. The biotin binding pockets made accessible again at lower pH, allowing the repetition of the synthesis and purification cycles. | ||
The assembly of precise tetrafunctional streptavidin conjugates relies on a peptide with an iminobiotin and 12 histidines, named the Ibio-His-tag (Fig. 1B). The Ibio-His-tag allows separating streptavidin (S) conjugates with different numbers of bound Ibio-His-tags on a Cu2+-NTA column using an imidazole elution gradient (S(Ibio-His-tag)1–4).17 Secondly, the pH-dependent binding of iminobiotin to streptavidin18 allows releasing the Ibio-His-tag from the conjugates by lowering the pH to 3.5 and linking a biotinylated molecules with precise stoichiometry. Previous reports have shown the preparation of monofunctional streptavidin conjugates with precise stoichiometries and different vacancies using this method.19
Repeating this chemistry over multiple cycles, we assemble in a step by step fashion up to four biotinylated molecules; in this case various fluorophores, on a single streptavidin with exact control over the stoichiometry (SA1B1C1D1, A: atto-565-biotin, B: atto-425-biotin, C: atto-665-biotin, D: biotin-5-fluorescein) (Fig. 1). To introduce the second biotinylated molecule, we started with a mono atto-565 labeled streptavidin, SA1, with three available biotin binding pockets, which was prepared as described previously.19 When SA1 (1 μM) was incubated with a second biotinylated molecule, atto-425-biotin (B, 2 μM) and the Ibio-His-tag (2 μM), a statistical mixture composed of SA1B3, S(Ibio-His-tag)1A1B2, S(Ibio-His-tag)2A1B1 and S(Ibio-His-tag)3A1 formed. Subsequently, the mixture separated on a Cu2+-NTA agarose column, where conjugate without the tag, SA1B3, was washed off and species bearing higher numbers of tag eluted at higher imidazole concentrations. In the chromatogram, three peaks with an absorbance typical for atto-565 (A) at 563 nm were observed, but only the first two absorbed at 436 nm typical for atto-425 (B) (Fig. 2A). Thus, the peaks with increasing imidazole concentration were assigned as S(Ibio-His-tag)1A1B2, S(Ibio-His-tag)2A1B1 and S(Ibio-His-tag)3A1, respectively.
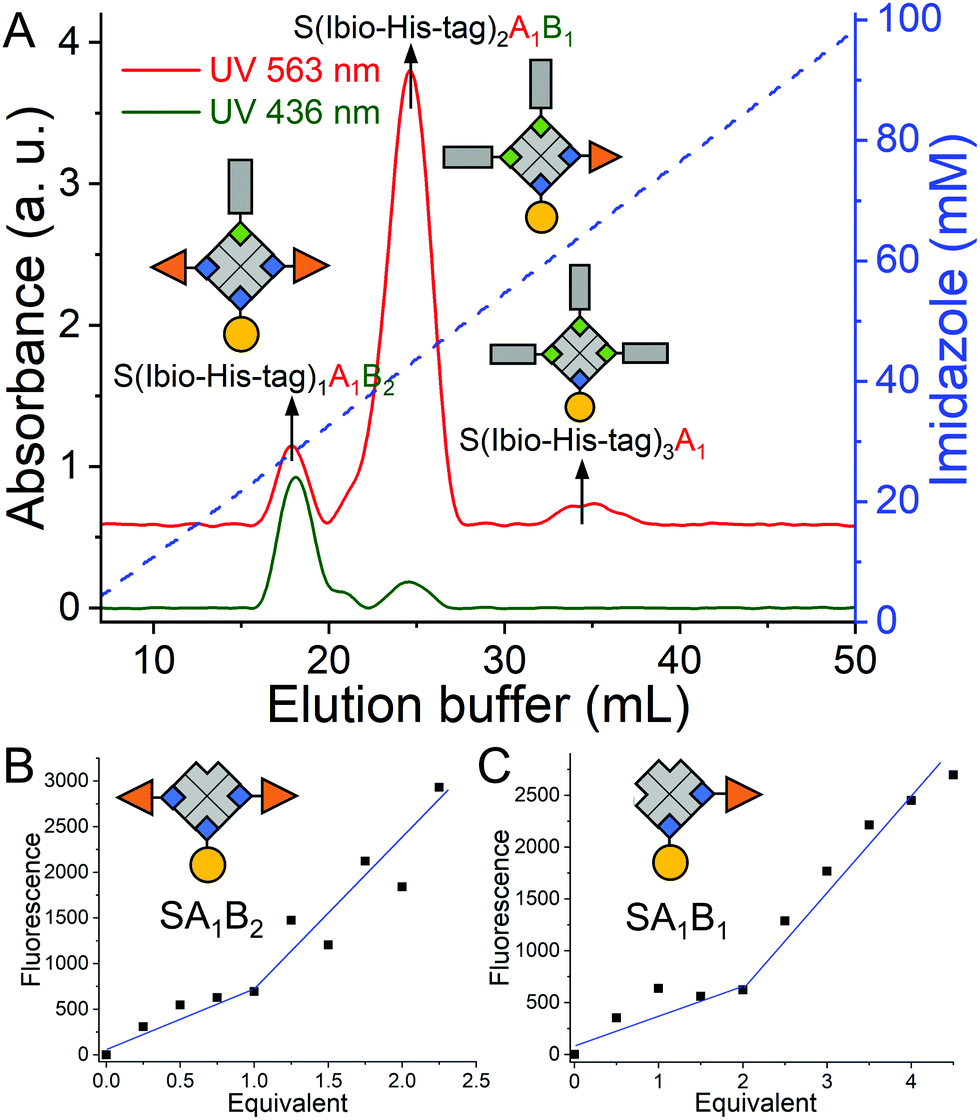 | ||
| Fig. 2 Preparation and characterization of difunctional streptavidin conjugates. (A) Chromatogram of the reaction mixture of SA1, atto-425-biotin and Ibio-His-tag. 1st peak: S(Ibio-His-tag)1A1B2 (27.8 mM imidazole, area 9.7%), 2nd peak: S(Ibio-His-tag)2A1B1 (43.6 mM imidazole, area 85.2%), 3rd peak: S(Ibio-His-tag)3A1 (63.2 mM imidazole, area 5.1%). SA1B3 (Area 2.5%) washed off without imidazole. The accessible biotin binding pockets of the species in the (B) 1st and (C) 2nd peak were titrated with biotin-5-fluorescein, A: atto-565-biotin, B: atto-425-biotin. | ||
To confirm the identity of the species, first the Ibio-His-tag was removed at lowered pH to yield well defined difunctionalized streptavidin conjugates with available biotin binding pockets, SA1B2 and SA1B1. Subsequently, the number of accessible biotin binding pockets was determined through the titration with biotin-5-fluorescein, which is quenched upon binding to streptavidin (Fig. 2B and C).20 The streptavidin conjugates in the first (SA1B2) and second (SA1B1) peak reacted with one and two equivalents of biotin-5-fluorescein, respectively.
The third biotinylated molecule was added onto the SA1B1 bioconjugate repeating the same protocol. Shortly, SA1B1 (A: atto-565-biotin, B: atto-425-biotin, 1 μM) was incubated with atto-665-biotin (C, 1.5 μM) and Ibio-His-tag (1.5 μM) and the reaction products were separated on a Cu2+-NTA agarose column. While the specie without the biotin tag, SA1B1C2, did not bind to the column, two species eluted at different imidazole concentrations (Fig. 3A). The first peak was assigned as S(Ibio-His-tag)1A1B1C1 because it only has one Ibio-His-tag and was the only species to absorb at 663 nm typical for atto-665 (C). The second peak at higher imidazole concentrations, was identified as S(Ibio-His-tag)2A1B1 since it has two tags.
 | ||
| Fig. 3 Preparation and characterization of tri- and tetra-functional streptavidin conjugates. (A) Chromatogram of the reaction mixture of SA1B1, atto-665-biotin and Ibio-His-tag. 1st peak: S(Ibio-His-tag)1A1B1C1 (34.1 mM imidazole, area 84.5%), 2nd peak: S(Ibio-His-tag)2A1B1 (41.5 mM imidazole, area 16.5%). SA1B1C2 (area 2.3%) washed off without imidazole. (B) SA1B1C1 required one equivalent of the biotin-5-fluorescein in the titration to saturate the biotin binding site, forming the tetrafunctional streptavidin conjugate, SA1B1C1D1. (C) Fluorescence emission spectrum (λEx = 400 nm) of the precise SA1B1C1 and a statistical mixture of streptavidin conjugates (S mixed with 1 equivalent of A, B and C). A: atto-565-biotin, B: atto-425-biotin, C: atto-665-biotin, D: biotin-5-fluorescein. | ||
Finally, to form an exact tetrafunctional streptavidin conjugate, SA1B1C1D1, it is sufficient to remove the Ibio-His-tag from S(Ibio-His-tag)1A1B1C1 at lowered pH and add one equivalent of the desired biotinylated molecule. When the above prepared SA1B1C1 was titrated with biotin-5-fluorescein (D), it reacted with one equivalent yielding SA1B1C1D1 (Fig. 3B). This experiment also confirms the structure of the SA1B1C1 conjugate with one open biotin binding pocket.
The importance of forming precise multifunctional streptavidins and not statistical mixtures was already apparent from the Förster resonance energy transfer (FRET) in SA1B1C1 (A: atto-565-biotin, B: atto-425-biotin, C: atto-665-biotin). When we excited the atto-425 at 400 nm, there were three emission peaks at 476, 594 and 690 nm for atto-425, atto-565 and atto-665, respectively due to their close proximity (Fig. 3C, Fig. S1, ESI†). On the other hand, when we formed a statistical mixture of conjugates by mixing one equivalent of each of the three fluorophores with streptavidin, there was only one peak at 476 nm upon excitation at 400 nm. Moreover, the characteristic UV-Vis absorbance peaks of the three fluorophores in SA1B1C1 support the stoichiometry of the complex (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B:C = 1.0:1.2:1.0) (Fig. S2, ESI†).
:B:C = 1.0:1.2:1.0) (Fig. S2, ESI†).
Next, we set out to combine four functionalities in one streptavidin conjugate to underline the potential for designing optimized cancer theranostics. For this purpose, we integrated the following functionalities within a single macromolecule: (i) atto-565-biotin (A) as a fluorescent label for tracking the fate of the conjugates, (ii) folic acid–PEG–biotin (F) for selective targeting of folic acid receptor positive cancer cells such triple negative breast cancer cells (MDA-MB-231),21 (iii) doxorubicin–biotin (D) (Fig. S3–S5, ESI†) as a broadly applied anti-cancer drug and (iv) a nucleus penetrating peptide–biotin (C) (Fig. S6 and S7, ESI†) for the targeted delivery of doxorubicin to the relevant subcellular compartment.22 Each of these biotinylated molecules was added sequentially according to the above order following the same method as described above (Fig. S8 and S9, ESI†). The yield of each step depends on the biotinylated molecules and was not the same as for the previous tetrafunctional conjugate.
Each of the biotinylated molecules within the tetrafunctional streptavidin conjugate should contribute to the overall function.
The folic acid in the conjugate is supposed to increase the selectivity for cancer cell types, which are folate receptor positive. To demonstrate this point, the uptake of streptavidin conjugates with (SA1F1, SA1F2 and SA1F3) and without folic acid (SA1) was evaluated in the folate receptor positive breast cancer cells, MDA-MB-231, and folate receptor-negative MCF-7 cells. After 4 h incubation the fluorescence signal from the atto-565 (A) showed that MDA-MB-231 cells incubated with different SAF conjugates were much brighter than MCF-7 cells (Fig. 4A and Fig. S10–S12, ESI†). Moreover, the uptake in MDA-MB-231 cells was clearly due to the folic acid as cells incubated with SA1 were not significantly fluorescent.
 | ||
| Fig. 4 (A) Confocal microscopy images of MDA-MB-231 (folate receptor positive) and MCF-7 (folate receptor negative) cells incubated with SA1F1 and MDA-MD-231 cells incubated with SA1. (B) MDA-MB-231 cells were incubated with 1 μM SA1F1, SA1F1D1 or SA1F1D1C1 in RPMI-1640 (no folic acid) medium for 4 hours. Atto-565 fluorescence shown in red and nuclei stained with DAPI are shown in blue. Scale bars are 25 μm. A: atto-565-biotin, F: folic acid–biotin, D: doxorubicin–biotin, C: nucleus penetrating peptide–biotin. | ||
Next, we evaluated the function of D (doxorubicin) and C (nucleus penetrating peptide) and their cooperativity in terms of the localization in the cells and cell toxicity. In particular, as doxorubicin stops DNA replication,23 we aimed to deliver it into the cell nucleus using this peptide. We found that the trifunctional streptavidin conjugate SA1F1D1 and tetrafunctional conjugate SA1F1D1C1 localized differently, when incubated with MDA-MB-231 cells (Fig. 4B). While SA1F1D1 localized in the cytoplasm and was excluded from the nucleus, SA1F1D1C1 with the nucleus penetrating peptide localized in the nucleus, as observed with confocal microscopy. This finding was further supported through the quantification of the fluorescence signal within the nucleus of the cells (Fig. S13, ESI†). Moreover, both SA1F1D1 and SA1F1D1C1 retained their selectivity for MDA-MB-231 cells over MCF-7 cells due to the folic acid modification (Fig. S11, ESI†).
Next, we tested the cell toxicity of the multifunctional streptavidin conjugates in MDA-MB-231 and MCF-7 cells. The cell viability as measured using the MTT test after 72 h, showed that the tetrafunctional SA1F1D1C1 was the most effective drug against the aggressive MDA-MB-231 cells (Fig. 5). In SA1F1D1C1 and SA1F1D1 the folic acid was responsible for the higher cell toxicity in MDA-MB-231 cells compared to MCF-7. While SA1F1D1C1 and SA1F1D1 had IC50 of 0.5 μM and 0.74 μM for MDA-MB-231 cells, respectively, their IC50 for MCF-7 cells were higher at 2 μM and 2.62 μM, respectively. This amounts to a fourfold selectivity for MDA-MB-231 cells over MCF-7. For comparison, pure doxorubicin had no specificity and was almost equally cell toxic to both cell types (Fig. S14, ESI†). In SA1F1D1C1, the nucleus penetrating peptide, C, increased the delivery of doxorubicin, D, to the cell nucleus, which contributes to the higher final efficacy. As a result, SA1F1D1C1 had a lower IC50 of 0.5 μM compared to SA1F1D1, which had a IC50 of 0.74 μM. The precise SA1F1D1 conjugate also outperformed the statistical mixtures of conjugates (S mixed with one equivalent of A, F, D and C). In fact, the SA1F1D1C1 was 1.5 times more cell toxic than the statistical mixture of conjugates (IC50 0.75 μM). Similarly, at 0.5 μM concentration MDA-MB-231 cells had a lower viability in the presence of SA1F1D1C1 (48%) than SA1F1D1 (57%) or the statistical mixture (57%) (Fig. S15, ESI†). Moreover, the toxicity of all three conjugates decreased for MDA-MB-231 cells in the presence of excess folic acid, but not for MCF-7 cells, which confirmed the role of the folic acid for cell selectivity.
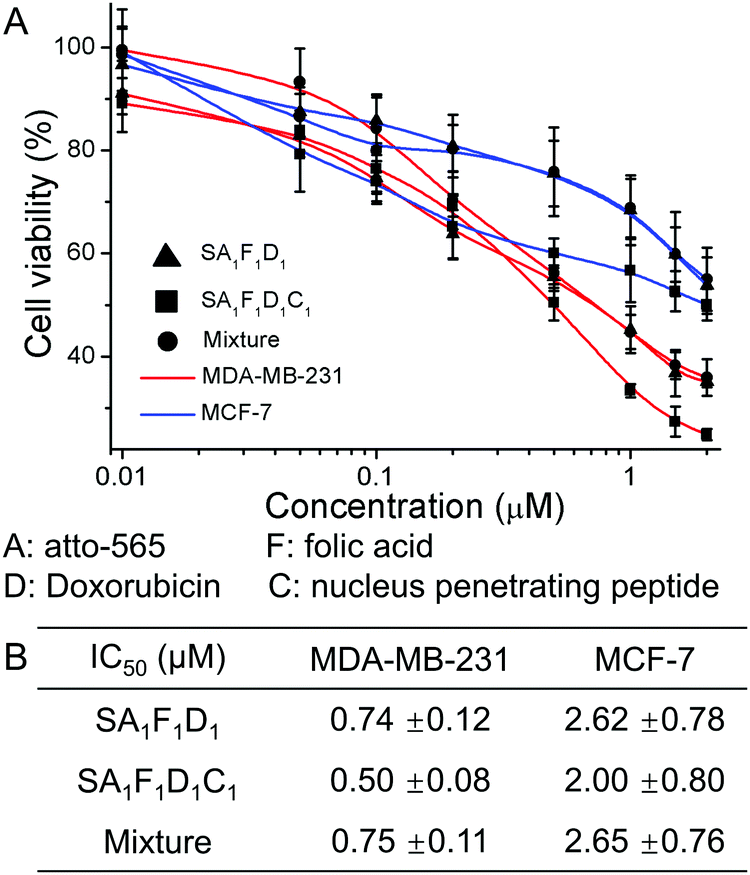 | ||
| Fig. 5 (A) Cell viability of MDA-MB-231 and MCF-7 cells were incubated with SA1F1D1, SA1F1D1C1 and a statistical mixture of streptavidin conjugates (S was mixed with one equivalent of A, F, D and C) for 72 hours. (B) Table of IC50 of different streptavidin conjugates. A: atto-565-biotin, F: folic acid–biotin, D: doxorubicin–biotin, C: nucleus penetrating peptide–biotin. | ||
In summary, we demonstrated how stoichiometrically precise tetrafunctional streptavidin conjugates can be prepared to bring together multiple functionalities. In particular, we demonstrated how the delivery of an anti-cancer drug can be improved through the combination of a fluorescent, a cell targeting group, a nucleus penetrating peptide and a cell toxic drug in a tetrafunctional streptavidin. The systematic introduction of each functionality allows studying its effect on viability, targeting and efficacy, which improves our understanding on the essential features of drug delivery agents and enables optimization. The precise tetrafunctional streptavidins also open the door to assembly combinatorial libraries of exact streptavidins taking advantage of a wide repertoire of biotinylated molecules (proteins, peptides, small molecules, lipids and nucleic acids). Thus, our study plays a significant role in expanding the streptavidin–biotin chemistry in biotechnology, nanostructure assemblies and drug delivery.
This work is part of the MaxSynBio consortium, funded by the Federal Ministry of Education and Research (BMBF) of Germany (FKZ 031A359L) and the Max Planck Society. D. X. thanks the Chinese Scholarship Council for a PhD fellowship. T. W., S. L. K. and A. H. thank the Deutsche Forschungsgemeinschaft (DFG), 316249678-SFB 1279 (C01) for funding.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Destito, R. Yeh, C. S. Rae, M. G. Finn and M. Manchester, Chem. Biol., 2007, 14, 1152–1162 CrossRef CAS PubMed.
- F. M. F. Santos, J. N. Rosa, N. R. Candeias, C. P. Carvalho, A. I. Matos, A. E. Ventura, H. F. Florindo, L. C. Silva, U. Pischel and P. M. P. Gois, Chem. – Eur. J., 2016, 22, 1631–1637 CrossRef CAS PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Goncalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew. Chem., Int. Ed., 2017, 56, 9346–9350 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- Y. Y. Kim, Y. Bang, A. H. Lee and Y. K. Song, ACS Nano, 2019, 13, 1183–1194 CrossRef CAS PubMed.
- M. Piffoux, A. K. A. Silva, C. Wilhelm, F. Gazeau and D. Tareste, ACS Nano, 2018, 12, 6830–6842 CrossRef CAS PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- S. Scarabelli, K. T. Tan, R. Griss, R. Hovius, P. L. D’Alessandro, T. Vorherr and K. Johnsson, ACS Sens., 2017, 2, 1191–1197 CrossRef CAS PubMed.
- L. Xue, Q. Yu, R. Griss, A. Schena and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 7112–7116 CrossRef CAS PubMed.
- R. Pasqualini and E. Ruoslahti, Nature, 1996, 380, 364–366 CrossRef CAS PubMed.
- N. M. Green, Methods Enzymol., 1990, 184, 51–67 CAS.
- M. Howarth, D. J. F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS PubMed.
- G. V. Dubacheva, C. Araya-Callis, A. G. Volbeda, M. Fairhead, J. Codee, M. Howarth and R. P. Richter, J. Am. Chem. Soc., 2017, 139, 4157–4167 CrossRef CAS PubMed.
- D. Kalderon, B. L. Roberts, W. D. Richardson and A. E. Smith, Cell, 1984, 39, 499–509 CrossRef CAS PubMed.
- V. Gaberc-Porekar and V. Menart, Chem. Eng. Technol., 2005, 28, 1306–1314 CrossRef CAS.
- S. L. Kuan, D. Y. W. Ng, Y. Z. Wu, C. Fortsch, H. Barth, M. Doroshenko, K. Koynov, C. Meier and T. Weil, J. Am. Chem. Soc., 2013, 135, 17254–17257 CrossRef CAS PubMed.
- D. Xu and S. V. Wegner, Chem. Sci., 2020, 11, 4422–4429 RSC.
- R. Mittal and M. P. Bruchez, Bioconjugate Chem., 2011, 22, 362–368 CrossRef CAS PubMed.
- Y. Ma, M. Sadoqi and J. Shao, Int. J. Pharm., 2012, 436, 25–31 CrossRef CAS PubMed.
- A. Eguchi, H. Furusawa, A. Yamamoto, T. Akuta, M. Hasegawa, Y. Okahata and M. Nakanishi, J. Controlled Release, 2005, 104, 507–519 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc04054a |
| This journal is © The Royal Society of Chemistry 2020 |