
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a porphyrin with histidine-like chelate: an efficient path towards molecular PDT/SPECT theranostics†
Steven Y.
Yap
 a,
Huguette
Savoie
a,
Isaline
Renard
bc,
Benjamin P.
Burke
bc,
Harry C.
Sample
a,
Saul
Michue-Seijas
a,
Stephen J.
Archibald
bc,
Ross W.
Boyle
*a and
Graeme J.
Stasiuk
*bd
a,
Huguette
Savoie
a,
Isaline
Renard
bc,
Benjamin P.
Burke
bc,
Harry C.
Sample
a,
Saul
Michue-Seijas
a,
Stephen J.
Archibald
bc,
Ross W.
Boyle
*a and
Graeme J.
Stasiuk
*bd
aDepartment of Chemistry and Biochemistry, Faculty of Science and Engineering, University of Hull, HU6 7RX, UK. E-mail: r.w.boyle@hull.ac.uk
bDepartment of Biomedical Sciences, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
cPositron Emission Tomography Research Centre, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
dDepartment of Imaging Chemistry and Biology, School of Biomedical Engineering and Imaging Sciences, King's College London, Fourth Floor Lambeth Wing, St Thomas’ Hospital, London SE1 7EH, UK. E-mail: graeme.stasiuk@kcl.ac.uk
First published on 12th August 2020
Abstract
The goal of “personalised” medicine has seen a growing interest in the development of theranostic agents. Bifunctional, and targeted-trifunctional, theranostic water-soluble porphyrins with a histidine-like chelating group have been synthesised via copper-catalysed azide–alkyne cycloaddition (CuAAC) “click” chemistry in high yield and purity. They are capable of photodynamic treatment and [99mTc(CO)3]+ complexation for single-photon emission computed tomography (SPECT) imaging, with a radiochemical yield of >95%. The toxicity and phototoxicity were evaluated on HT-29 cells, DU145, and DU145-PSMA cell lines, with the targeted theranostic showing more potent phototoxicity towards DU145-PSMA expressing cells.
Theranostics, a term given to the combination of therapy (thera-) and diagnostic (-nostic) modalities into a single construct or molecule, has seen a growing interest in the field of medicine.1,2 Conventional treatments often involve a “one-size-fits-all” technique, and can have significant differences in the treatment outcomes between patients. Theranostics naturally lends itself towards the goal of “personalised” medicine, capable of optimising and improving treatment outcomes.1
Porphyrins are excellent candidates for molecular theranostic agents. There are multiple studies documenting their use in medicine, especially as a photosensitiser in the field of photodynamic therapy (PDT).1,2 Porphyrins, when combined with molecular oxygen and visible light (400–700 nm), are capable of delivering potent and selective photodynamic treatment to tumour tissues.1,3–5 By combining a diagnostic modality with a PDT drug, this allows the careful monitoring of the accumulation of PDT drug within the body, facilitating the tuning of treatment by light irradiation when the accumulation of drug is at a maximum, ultimately improving treatment outcomes.
Single-photon emission computed tomography (SPECT) is a highly sensitive imaging technique that requires the administration of a radiotracer. SPECT requires the radiotracer to biodistribute/accumulate in the body. Images are then obtained using a gamma camera and computed to provide a 3D image of the location of the radiotracer in the body. 99mTc is often used in SPECT imaging, since it has desirable nuclear properties with a half-life, t1/2, of 6 hours and a main gamma emission, Eγ, at 140 keV (89% emission).6 Its half-life is long enough to manipulate the isotope to prepare various radiopharmaceuticals and its gamma emission allows established chemistry. Its availability from a 99Mo/99mTc generator and relative low cost, have made it the most commonly used radioisotope in diagnostic nuclear medicine.6
Schibli has shown that alkyne/azide-modified amino acids “click”, via copper-catalyse azide alkyne cycloaddition (CuAAC), with other azide/alkyne-functionalised molecules, to form a histidine-like structure that can efficiently complex [99mTc(CO)3]+.7 The utilisation of “click” chemistry unlocks the possibility to use a high yielding and high purity “click” reaction to combine two moieties together, opening the way towards molecular SPECT theranostic medicine.
Despite the potential utility of 99mTc-labelled porphyrins, there are limited reports on the combination of PDT with SPECT,8–13 with only one example on the usage of conjugated histidine-like structures as a 99mTc chelate.13 Herein, following our interest in developing porphyrin molecular theranostics,14–17 we report the synthesis of two water-soluble porphyrins conjugated with histidine-like ligands using CuAAC “click” chemistry in high yields and with minimal impurities, and show that they are capable of complexing [99mTc(CO)3]+ with good radiochemical yields.
Initially, a water-soluble azide-appended porphyrin 1 was synthesised via 5 steps as previously reported (detail in ESI†).18
Commercially available D-propargylglycine was conjugated with porphyrin 1 using CuAAC (Scheme 1).19 With excess D-propargylglycine, quantitative conversion of porphyrin 1 to conjugate 2 was shown using HPLC analysis, with a clear shift in HPLC retention time from 10.8 min to 7.5 min (Fig. S1A, ESI†). Subsequently, purification of conjugate 2 was carried out using a counter-ion exchange technique previously reported by our group.14 Briefly, the solubility of the porphyrin can be controlled by exchanging the anionic counter-ions. Exchange of the iodide anions with hexafluorophosphate rendered the conjugate insoluble in water. This allowed filtration and removal by washing of excess starting materials and reagents. Subsequent anionic conversion to chloride yielded the water-soluble conjugate 2. The identity of conjugate 2 was further confirmed using NMR and mass spectrometry.
 | ||
Scheme 1 Synthesis of conjugate 2 and conjugate 3 and the subsequent complexation of natural rhenium and 99mTc. (i) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 t-butanol:water, D-propargylglycine, CuSO4, sodium ascorbate, microwave (20 min, 75 W, 50 °C). (ii) 6, 1:1 t-butanol:water, CuSO4, sodium ascorbate, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, microwave (3 hours, 75 W, 70 °C). (iii) water, LiOH, room temperature 3 hours. (iv) phosphate buffer pH 7, [Re(CO)3Br3][NEt4]2, 65 °C, 2 hours. (v) [99mTc(CO)3(H2O)3]+, pH 7.4, 90 °C, 30 min. :1 t-butanol:water, D-propargylglycine, CuSO4, sodium ascorbate, microwave (20 min, 75 W, 50 °C). (ii) 6, 1:1 t-butanol:water, CuSO4, sodium ascorbate, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, microwave (3 hours, 75 W, 70 °C). (iii) water, LiOH, room temperature 3 hours. (iv) phosphate buffer pH 7, [Re(CO)3Br3][NEt4]2, 65 °C, 2 hours. (v) [99mTc(CO)3(H2O)3]+, pH 7.4, 90 °C, 30 min. | ||
We envisaged introduction of an additional targeted functionality could be facilitated by using lysine, instead of glycine, which has an amino group side chain. However, there is no commercially available propargyl lysine, hence, modification of commercially available N(ε)-Boc-Lys(OMe) was required. Scheme 2 shows the modification of N(ε)-Boc-Lys(OMe) to introduce an alkyne-functionality and also a conjugatable linker, 5. This linker allows further conjugation with a targeting moiety. We have chosen a well-described small molecule dipeptide that acts as an inhibitor against prostate specific membrane antigen (PSMA), which is overexpressed in metastatic prostate cancer.20 The targeting dipeptide has the structure: lysine-urea-glutamate, with the glutamate moiety responsible for targeting and binding to the pharmacophore pocket of PSMA.21,22 Peptide conjugation and subsequent deprotection of tert-butyl groups using trifluoroacetic acid (TFA) yielded the clickable 6 capable of targeting PSMA.
 | ||
| Scheme 2 Synthesis of trifunctional-propargyl lysine. (i) DMF, K2CO3, propargyl bromide, room temperature, 48 hours. (ii) DCM, TFA, room temperature, 2 hours. (iii) DMF, disuccinimidyl suberate, triethylamine, room temperature, overnight. (iv) DMF, triethylamine, PSMA-dipeptide inhibitor, room temperature, overnight. (V) DCM, TFA, room temperature, 3 hours. | ||
Using similar CuAAC reaction conditions for the synthesis of conjugate 2, quantitative conversion of porphyrin 1 to 3 was observed, with a clear shift in HPLC retention time from 10.8 to 8.6 min (Fig. S1B, ESI†). Using the same counter-ion exchange technique, 3 was purified, isolated, and characterised. Deprotection of methyl ester on 3 was carried out in aqueous LiOH yielding trifunctional water-soluble conjugate 4 in quantitative yield.
Chemically and physically similar rhenium was complexed with conjugate 2 and 4 (Scheme 1).23,24 Rhenium is a commonly used surrogate for technetium as there is no stable technetium isotopes.25 In addition, the availability of β-particle emitting rhenium 186/188 isotopes further expand the utility of 2 and 4 as radiotherapeutic agent.26 Complexation with rhenium was carried out to produce non-radioactive complexes for use as HPLC standards and in biological cytotoxicity evaluations. With a slight excess of rhenium at 65 °C for 2 hours, HPLC analysis of the reaction mixture showed quantitative conversion of conjugates 2 and 4 to their corresponding complexes, [Re(2)] and [Re(4)], exhibited by a clear shift in HPLC with retention times of 10.2 min and 10.8 min, respectively (Fig. S1, ESI†). 13C NMR spectroscopy showed the addition of three carbonyl carbon peaks between δ 197–199 for both complexes, [Re(2)] and [Re(4)]. High resolution mass spectrometry of [Re(2)] and [Re(4)] also gave their corresponding mass peak, giving evidence for the successful complexation of rhenium.
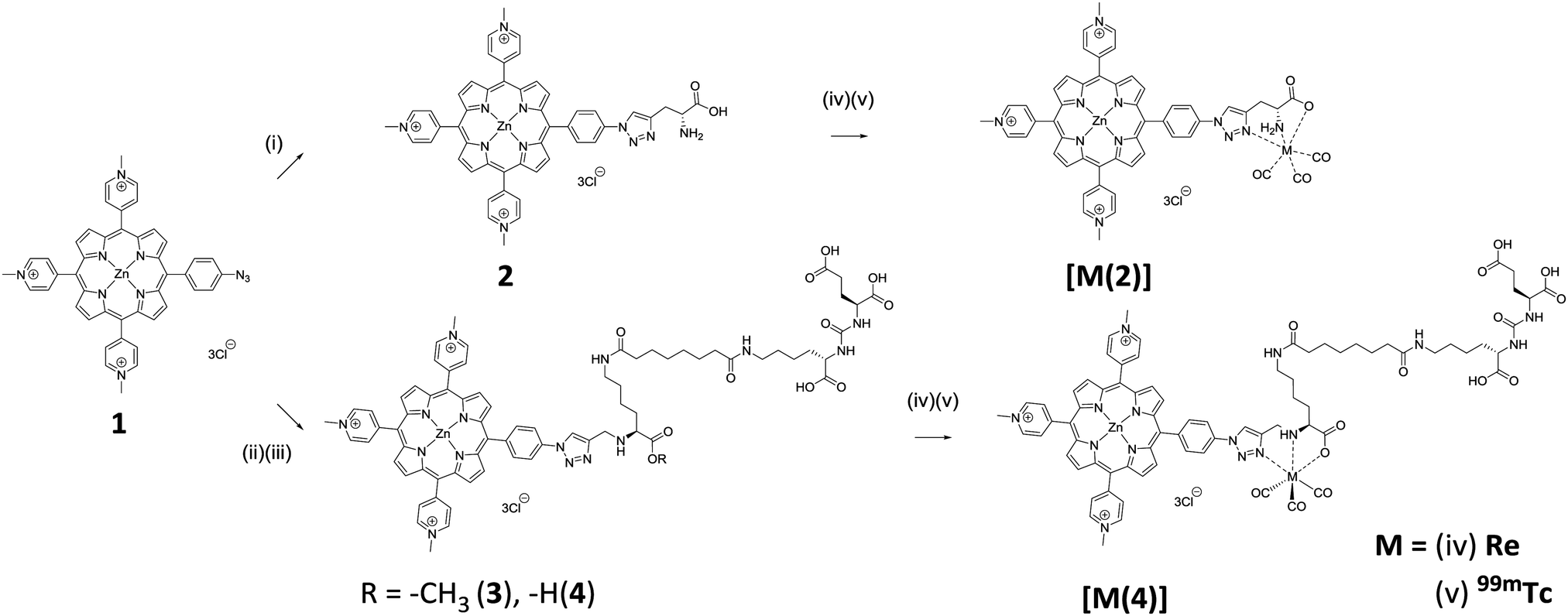
Successful synthesis of conjugate 2 and 4 and their corresponding rhenium complexes prompted the radiolabelling with 99mTc. Firstly, [99mTc(CO)3(H2O)3]+ was prepared according to the literature method (Fig. S2 and S3, ESI†).27 After careful optimisation of reaction conditions–reaction time, pH, reaction temperature, and conjugate concentration, both conjugate 2 and 4 can be produced in >95% radiochemical yield (RCY) at a concentration of 150 μM at pH 7.4, 99 °C for 30 min, as determined using radio-HPLC. Radio-HPLC analysis showed the formation of a single radiolabelled product which matches well with the retention time of the rhenium complexes (Fig. 1), giving evidence for the formation of the desired radiolabelled complexes, [99mTc(2)] and [99mTc(4)]. With these optimised radiolabelling conditions, specific activities of 76 GBq μmol−1 and 96 GBq μmol−1 were achieved for conjugates 2 and 4, respectively. The lipophilicity of the tracers were determined using the shake-flask method and were found to be −0.57 ± 0.08 and −1.31 ± 0.01 for [99mTc(2)] and [99mTc(4)], respectively. This indicates [99mTc(4)] is more hydrophilic due to the presence of extra carboxylate groups.
 | ||
| Fig. 1 Overlays of radio-HPLC of [99mTc(2)] and [99mTc(4)] and UV-vis HPLC chromatograms [Re(2)] and [Re(4)]. Reaction conditions: [L] = 150 μM, t = 30 min, T = 99 °C, pH 7.4. | ||
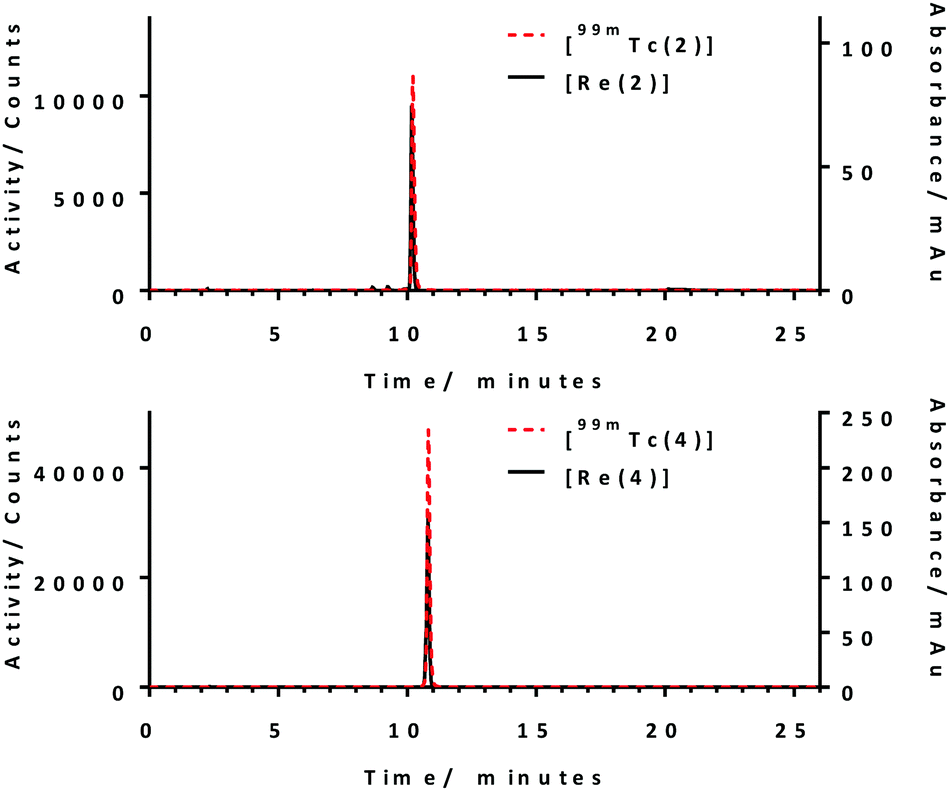
The stability of the radiolabelled complexes, [99mTc(2)] and [99mTc(4)], were tested against 50% foetal bovine serum (FBS) with an activity of 35–40 MBq (at concentration of 460–530 nM for [99mTc(2)] and 360–420 nM for [99mTc(4)]). [99mTc(2)] shows 94% of 99mTc still associated and [99mTc(4)] shows remarkable stability against 50% FBS with >99% 99mTc still associated after incubation for 6 hours (Fig. 2). The high serum stability of these 99mTc conjugates was expected as literature has shown the stability of histidine and histidine-like complexes against serum.7,25,28
 | ||
| Fig. 2 Serum stability of radiolabelled conjugates, [99mTc(2)] and [99mTc(4)]. | ||
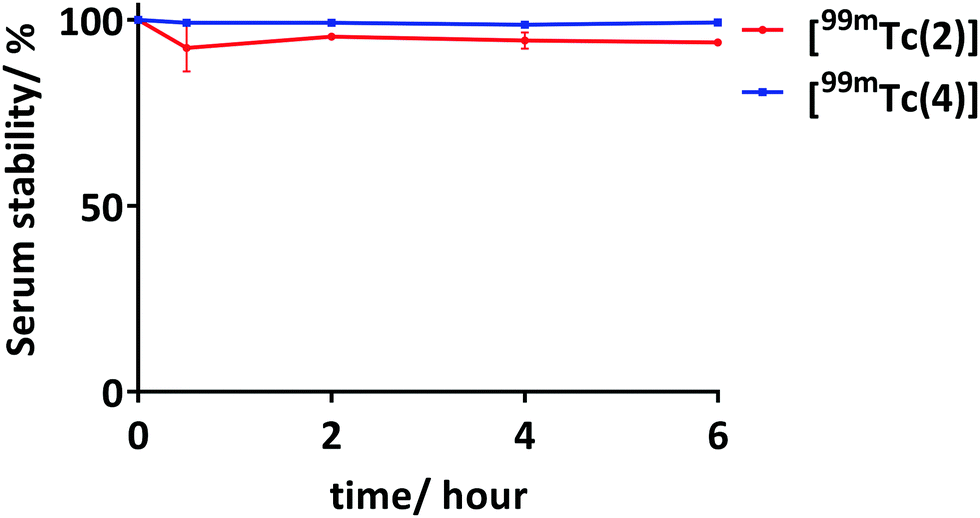
Toxicity and phototoxicity of 2 and [Re(2)] were evaluated on human adenocarcinoma (HT-29) cells. Cells were incubated with 2 and [Re(2)] at varying concentrations (1–18 μM) for 1 hour and irradiation was carried out using a constant dose of visible light (20 J cm−2; 400–700 nm). The results obtained were compared with a non-irradiated control. Under these conditions, both conjugate 2 and [Re(2)] were capable of inducing >50% cell death at 4.5 μM with minimal dark toxicity observed in the non-irradiated controls (Fig. 3 and Fig. S6, ESI†).
 | ||
| Fig. 3 Percentage cell survival of HT-29 (top) and DU145 and DU145-PSMA (botom), irradiated (red) and non-irradiated (black) cells, as determined using MTT assay. Cells were incubated with varying concentration of [Re(2)] (top) and [Re(4)] (bottom) and irradiated cells received 20 J cm−2 white light. MTT assays were carried out in three quadruplicates. | ||
As 4 and [Re(4)] have a targeting molecule which inhibits PSMA, two different human prostate cancer cell lines were used to evaluate the toxicity and phototoxicity of 4 and [Re(4)], namely DU145, which does not express PSMA, and DU145-PSMA, a PSMA-expressing variant.29 Initially, the level of PSMA expression on both cell lines was determined by flow cytometry analysis (Fig. S5, ESI†). Varying concentration of 4 and [Re(4)] (1.5–24 μM) were incubated with these cell lines for 30 min prior to visible light irradiation. Results obtained were also compared with non-irradiated controls (Fig. 3 and Fig. S6, ESI†). Without irradiation, both 4 and [Re(4)] show minimal toxicity at these concentration with more than 86% cell survival in both DU145 and DU145-PSMA. Under irradiation with visible light, both 4 and [Re(4)] were capable of inducing death in both DU145 and DU145-PSMA cells, with more potent phototoxicity towards DU145-PSMA. At a concentration of 6 μM, 85% cell survival was observed for 4 in DU145 cells with light irradiation, while only 17% cell survival was observed in DU145-PSMA cells under similar conditions. Similarly, [Re(4)] showed minimal phototoxicity towards DU145 cells at a concentration of 1.5 μM with >90% cell survival, but was capable of inducing near 70% cell death in DU145-PSMA under similar conditions. These results show, 4 and [Re(4)] are capable of inducing greater cell death in DU145-PSMA cells compared to DU145 cells under similar conditions. This is due to the overexpression of PSMA on DU145-PSMA, resulting in the higher binding/uptake of 4 and [Re(4)] into DU145-PSMA.
Nevertheless, phototoxicity of 4 and [Re(4)] towards native DU145 increases with concentration (Fig. 3 and Fig. S6, ESI†). This could be due to a combination of low level native expression of PSMA on DU145 and non-specific uptake/binding of 4 and [Re(4)].
In conclusion, the synthesis and evaluation of [99mTc(2)] and [99mTc(4)] show the validity of a click to chelate method to bifunctional theranostic and trifunctional targeted theranostic agents. Excellent radiochemical yields (>95%) were achieved for radiolabelling of both 2 and 4. Individually, 2, [Re(2)], 4, and [Re(4)] were capable of inducing cell death (>90%) when irradiated with light while showing minimal toxicity in the absence of light. Specificity of 4 and [Re(4)] towards PSMA was confirmed as more cell death was induced in DU145-PSMA when compared to the DU145 under similar conditions. The utilisation of CuAAC “click” chemistry demonstrated an effective strategy for combining different moieties into a single molecule, providing a route towards multifunctional 99mTc labelled porphyrin PDT/SPECT theranostic agents. This strategy will allow the possibility of molecular multifunctional porphyrin agents to be applied in in vivo and clinical settings.
We gratefully acknowledge Dr Assem Allam and his family for the generous donation to help found the PET Research Centre at the University of Hull. Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry at Swansea University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916–966 CrossRef CAS PubMed.
- Y. Zhang and J. F. Lovell, Theranostics, 2012, 2, 905–915 CrossRef CAS PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- I. J. Macdonald and T. J. Dougherty, J. Porphyr. Phthalocyanines, 2001, 5, 105–129 CrossRef CAS.
- S. Jurisson, D. Berning, W. Jia and D. Ma, Chem. Rev., 1993, 93, 1137–1156 CrossRef CAS.
- T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwé and R. Schibli, J. Am. Chem. Soc., 2006, 128, 15096–15097 CrossRef CAS PubMed.
- M. Tsutsui and C. p. Hrung, Chem. Lett., 1973, 941–942 CrossRef CAS.
- P. M. Santos, M. Laranjo, A. C. Serra, A. M. Abrantes, M. Piñeiro, J. Casalta-Lopes, D. Trindade, J. Maia, A. Rocha-Gonsalves and M. F. Botelho, J. Label. Compd. Radiopharm, 2014, 57, 141–147 CrossRef CAS PubMed.
- M. Subbarayan, S. J. Shetty, T. S. Srivastava, O. P. D. Noronha and A. M. Samuel, J. Porphyr. Phthalocyanines, 2001, 5, 824–828 CrossRef CAS.
- C. Spagnul, R. Alberto, G. Gasser, S. Ferrari, V. Pierroz, A. Bergamo, T. Gianferrara and E. Alessio, J. Inorg. Biochem., 2013, 122, 57–65 CrossRef CAS PubMed.
- T. Gianferrara, C. Spagnul, R. Alberto, G. Gasser, S. Ferrari, V. Pierroz, A. Bergamo and E. Alessio, ChemMedChem, 2014, 9, 1231–1237 CrossRef CAS PubMed.
- Y. Liu, B. Shen, F. Liu, B. Zhang, T. Chu, J. Bai and S. Bao, Nucl. Med. Biol., 2012, 39, 579–585 CrossRef CAS PubMed.
- S. Y. Yap, T. W. Price, H. Savoie, R. W. Boyle and G. J. Stasiuk, Chem. Commun., 2018, 54, 7952–7954 RSC.
- F. Bryden, H. Savoie, E. V. Rosca and R. W. Boyle, Dalton Trans., 2015, 44, 4925–4932 RSC.
- G. M. Entract, F. Bryden, J. Domarkas, H. Savoie, L. Allott, S. J. Archibald, C. Cawthorne and R. W. Boyle, Mol. Pharm., 2015, 12, 4414–4423 CrossRef CAS PubMed.
- T. W. Price, S. Y. Yap, R. Gillet, H. Savoie, L. J. Charbonnière, R. W. Boyle, A. M. Nonat and G. J. Stasiuk, Chem. – Eur. J., 2020, 26, 7602–7608 CrossRef CAS PubMed.
- F. Bryden, A. Maruani, H. Savoie, V. Chudasama, M. E. B. Smith, S. Caddick and R. W. Boyle, Bioconjugate Chem., 2014, 25, 611–617 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- M. Felber, M. Bauwens, J. M. Mateos, S. Imstepf, F. M. Mottaghy and R. Alberto, Chem. – Eur. J., 2015, 21, 6090–6099 CrossRef CAS PubMed.
- C. Bařinka, M. Rovenská, P. Mlčochová, K. Hlouchová, A. Plechanovová, P. Majer, T. Tsukamoto, B. S. Slusher, J. Konvalinka and J. Lubkowski, J. Med. Chem., 2007, 50, 3267–3273 CrossRef PubMed.
- World Intellectual Property Organization, WO2008058192A2, 2008.
- T. Mindt, H. Struthers, E. Garcia-Garayoa, D. Desbouis and R. Schibli, Chim. Int. J. Chem., 2007, 61, 725–731 CrossRef CAS.
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 RSC.
- H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, Chem. – Eur. J., 2008, 14, 6173–6183 CrossRef CAS PubMed.
- C. A. Kluba and T. L. Mindt, Molecules, 2013, 18, 3206–3226 CrossRef CAS PubMed.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135–3136 CrossRef CAS PubMed.
- R. Schibli, R. La Bella, R. Alberto, E. Garcia-Garayoa, K. Ortner, U. Abram and P. A. Schubiger, Bioconjugate Chem., 2000, 11, 345–351 CrossRef CAS PubMed.
- F. Kampmeier, J. D. Williams, J. Maher, G. E. Mullen and P. J. Blower, EJNMMI Res., 2014, 4, 13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and spectras. See DOI: 10.1039/d0cc03958f |
| This journal is © The Royal Society of Chemistry 2020 |