
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bis(germylene) functionalized metal-coordinated polyphosphide and its isomerization†
Ravi
Yadav
 a,
Bhupendra
Goswami
a,
Thomas
Simler
a,
Christoph
Schoo
a,
Stephan
Reichl
b,
Manfred
Scheer
b and
Peter W.
Roesky
*a
a,
Bhupendra
Goswami
a,
Thomas
Simler
a,
Christoph
Schoo
a,
Stephan
Reichl
b,
Manfred
Scheer
b and
Peter W.
Roesky
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute of Inorganic Chemistry, University of Regensburg, Universitätsstraße 31, 93040 Regensburg, Germany
First published on 4th August 2020
Abstract
Pentaphosphaferrocene, [Cp*Fe(η5-P5)] (Cp* = η5-C5Me5), was used as a polyphosphorus source to obtain germylene–polyphosphide complexes. A stepwise reactivity was observed between the di-germylene, [LGe–GeL] (L = {PhC(NtBu)2}), and [Cp*Fe(η5-P5)]. Firstly, reductive homolytic cleavage of the Ge–Ge single bond in [LGe–GeL] led to [(LGe)2{(μ,η4-P5)FeCp*}]. This complex showed an unprecedented isomerization.
Phosphorus containing organo-heterocyclic compounds are very well documented1 and have found applications in material science,2 pharmaceutical industries,3 and ligand design for coordination chemistry.4 However, phosphorus heterocyclic compounds based on heavier group-14 elements are relatively scarce.5 Recently, direct functionalization of white phosphorus with low-valent species to access phosphorus heterocycles has attracted wide-spread attention.6 Low-valent main group compounds are usually highly susceptible towards redox reactions and have been efficiently utilized to reduce white phosphorus and generate phosphorus containing heterocyclic compounds.6a,7 For example, the reactions of different carbenes with white phosphorus resulted in organo-phosphorus cages, chains, and rings.8 Similarly, the reaction of white phosphorus with silylenes furnished sila-phospha cages and rings.7c,9 Also, low-valent germanium compounds have been reported to react with P4 leading to germa-phospha heterocyclic compounds.10
The isolobality between [CH] and [P] fragments led to the prediction that the [cyclo-P5]− moiety may also be used to make sandwich compounds similar to ferrocene.11 In 1987, Scherer and co-workers synthesized pentaphosphaferrocene, [Cp*Fe(η5-P5)], in an elegant approach by co-thermolysis of white phosphorus and [Cp*Fe(CO)2]2.12 [Cp*Fe(η5-P5)] has revealed to be a very useful metalloligand for further coordination chemistry due to the presence of a C5 symmetric arrangement of lone pairs on the cyclo-P5 ring. By using [Cp*Fe(η5-P5)], one of us has carried out pioneering work in the field of inorganic supramolecular and polymer chemistry.13 Notably, the redox chemistry of [Cp*Fe(η5-P5)] is different from that of ferrocene, due to the involvement of the cyclo-P5 ring in redox events.14 Also, the reactivity of [Cp*Fe(η5-P5)] towards main group nucleophiles and lanthanides has been investigated.15 Very recently, we have shown that [Cp*Fe(η5-P5)] could be used as an air-stable polyphosphorus source to obtain silicon- or aluminium-polyphosphorus heterocyclic compounds.16
Interestingly, the reaction of [Cp*Fe(η5-P5)] with different silylenes, e.g. [LSiCl],17 [LSi{N(SiMe3)2}],18 and [LSi–SiL]19 resulted in different reaction pathways such as substitution A, addition B, and ring expansion C reactions, respectively (Chart 1).16a Such a rich reactivity of [Cp*Fe(η5-P5)] with species featuring low-valent silicon prompted the question as to whether a similar reactivity would be observed with the heavier germylene analogues [LGeCl] and [LGe–GeL].20 Low-valent Si species are prone to become Si(IV) via oxidation, however, this pathway is not obvious for low-valent Ge compounds, which may result in a different reactivity.21 Herein, we report on the reactivity of [Cp*Fe(η5-P5)] with these germylenes and the isolation of the first example of a bis(germylene)-functionalized transition-metal polyphosphide complex. Furthermore, the isolated bis(germylene)–polyphosphide complex revealed an unprecedented constitutional isomerization.
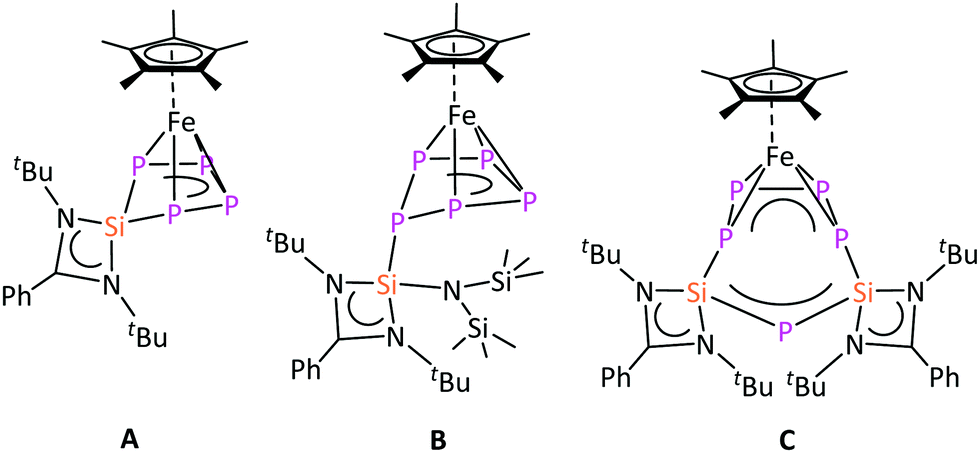 | ||
| Chart 1 Selected products obtained from the reaction of [Cp*Fe(η5-P5)] with different silylenes.16a | ||
In contrast to [LSiCl], the heavier analogue of amidinate chloro-silylene, [LGeCl], does not react with [Cp*Fe(η5-P5)] even after heating the reaction mixture at 80 °C for 24 h (Scheme 1), as evidenced by 1H NMR spectroscopy (Fig. S2, ESI†). This lack of reactivity can be explained by the lower oxidation potential (in absolute values) of [LGeCl] compared to [LSiCl]. Since the di-silylene [LSi–SiL] was found to be more reactive towards [Cp*Fe(η5-P5)] than [LSiCl], as the Si atoms are formally in the +1 oxidation state in [LSi–SiL],16a the corresponding di-germylene, [LGe–GeL], was employed for further investigations. The lower reactivity of [LGe–GeL] compared to [LSi–SiL] could be advantageously utilized to observe some intermediates that were anticipated to be also formed in the reaction with [LSi–SiL], but could not be identified. The reaction between [LGe–GeL] and [Cp*Fe(η5-P5)] at room temperature in toluene for 12 hours led to a mixture of products, as monitored by 1H and 31P{1H} NMR studies. Interestingly, the intensity of the different resonances in the 1H NMR spectra changed over time. Besides the starting material, [Cp*Fe(η5-P5)], some new compounds were formed. As outlined in the following sections, two of these compounds could be successfully identified.
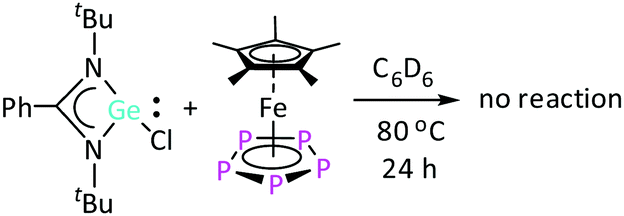 | ||
| Scheme 1 Reaction attempt between [LGeCl] and [Cp*Fe(η5-P5)]. | ||
The relatively slow conversion observed when using [LGe–GeL] prompted us to follow the reaction between [LGe–GeL] and [Cp*Fe(η5-P5)] by starting the reaction at −78 °C, warming up to room temperature and stirring for half an hour (route A; Scheme 2). After work-up, crystals of [(LGe)2{(μ,η4-P5)FeCp*}] (1) were obtained in 60% yield. Interestingly, the same product 1 was formed in quantitative yield in the reaction of [{K(dme)}2Cp*Fe(η5-P5)]14 with 2 equivalents of [LGeCl] by salt elimination (route B, Scheme 2). The 1H NMR spectrum of complex 1 showed two singlets at δ = 1.23 and 1.32 ppm corresponding to the four tBu groups on the amidinate ligands, indicating different environments for the two [LGe] moieties. The resonance corresponding to the methyl protons of the Cp* group is downfield shifted to δ = 1.90 ppm compared to δ = 1.08 ppm in [Cp*Fe(η5-P5)]. The 31P{1H} NMR spectrum showed an AMM′XX′ spin system corresponding to magnetically non-equivalent P atoms with chemical shifts at δ = −45.7 (PXX′), 43.5 (PMM′), and 150.5 (PA) ppm (Fig. 1), indicating the formation of an envelope shape for the cyclo-P5 ring. The different JPP coupling constants could be determined by iterative line fitting of the 31P{1H} NMR spectrum (Section 4.1, ESI†) and are in the usual range of those observed for complexes containing an envelope-shaped cyclo-P5 ring.16
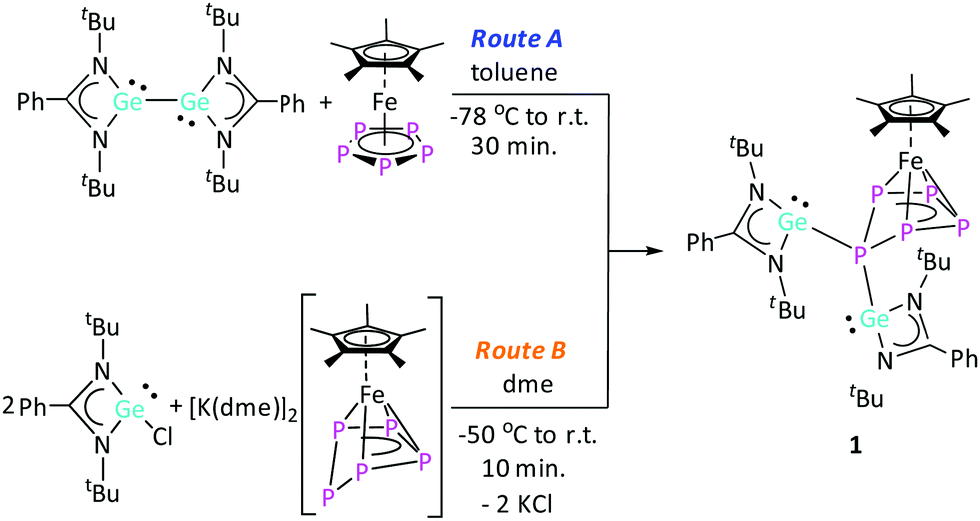 | ||
| Scheme 2 Synthesis of complex 1. | ||
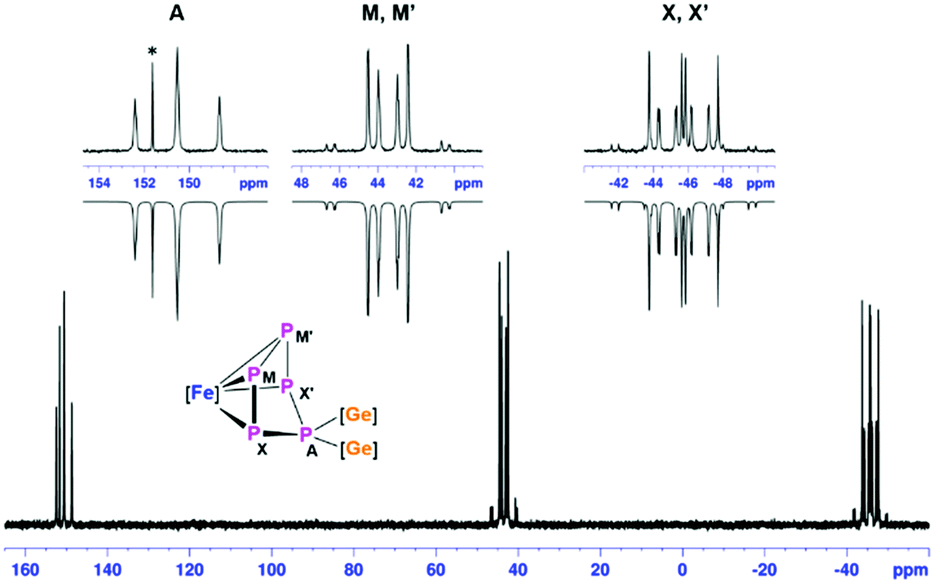 | ||
| Fig. 1 31P{1H} NMR spectrum at 298 K of compound 1 in C6D6 with nuclei assigned to an AMM′XX′ spin system; insets: extended signals (upward) and simulations (downward). See ESI† for details. | ||
The molecular structure of complex 1 further confirmed the formation of an envelope-shaped cyclo-P5 ring η4-coordinated to the [Cp*Fe]+ fragment (Fig. 2). The Fe–P bond distances (2.311(2)–2.357(2) Å) are in the usual range for similar compounds featuring an envelope-shaped cyclo-P5.14,15 The Ge1–P1 (2.4394(14) Å) and Ge2–P1 (2.4978(13) Å) bond lengths are similar to those in other tri-coordinated germylenes in a polyphosphide environment, for example, [RGe–(μ-P2)–GeR]22 (2.439(1) Å) (R = [(p-tolyl)2B{1-(1-adamantyl)-3-yl-2-ylidene}2]) and [P7Ge(N(SiMe3)2)]2− (2.504(1) and 2.526(1) Å).23 The angles between the N–Ge–P1 atoms, from 94.57(12)° to 97.36(12)°, are in the typical range for tri-coordinated germylenes.24 The two germylene moieties, [LGe]+, in complex 1 are in a trans-conformation with respect to the orientation of the lone pairs on the Ge centres. Complex 1 results from the homolytic cleavage of the Ge–Ge single bond in [LGe–GeL] and the two-electron reduction of [Cp*Fe(η5-P5)], and corresponds to a rare example of a germylene in the coordination sphere of a polyphosphide.22,23,25 Furthermore, to the best of our knowledge, complex 1 is the first example of a bis(germylene) functionalized transition-metal polyphosphide.26
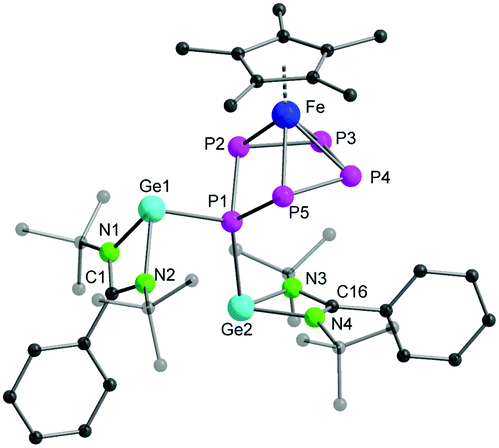 | ||
| Fig. 2 The molecular structure of complex 1 in the solid state. The hydrogen atoms and the solvent molecule in the unit cell are omitted for clarity. Bond lengths and angles are given in ESI.† | ||
Interestingly, [LGe–GeL] is reacting with exclusively one equivalent of [Cp*Fe(η5-P5)], even when the reaction is carried out in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, respectively. In this case, complex 1 together with one equivalent of unreacted [Cp*Fe(η5-P5)] are obtained. It should be noted that complex 1 is not stable in solution at room temperature and further reacts slowly. However, by careful control of the reaction time, complex 1 could be isolated in a pure form (details in ESI†). In order to identify the subsequent product(s) of the degradation of complex 1, an NMR study of complex 1 was carried out. The 1H NMR spectrum showed the conversion of 1 into new species (vide infra) along with re-formation of a small amount of [Cp*Fe(η5-P5)]. However, the rate of the conversion at room temperature was found to be very slow, which prevented full conversion, even after one month (Fig. S8 and S9, ESI†). Therefore, to increase the rate, complex 1 was heated at 40 °C for 48 h (Fig. S10, ESI†). On a preparative scale, the reaction between [LGe–GeL] and [Cp*Fe(η5-P5)] was performed in toluene at 40 °C for 24 h. As a result, after work-up, a mixture of crystals was obtained, consisting mostly of 1 and [Cp*Fe(η5-P5)] along with a few brown-coloured crystals of [(LGe){(μ,η3-P5)(η1-GeL)FeCp*}] (2), which were identified by single crystal X-ray diffraction studies (for 2cf.Scheme 3 and Fig. 3). Also, attempts to obtain pure complex 2 from isolated crystalline complex 1 in toluene at 40 °C resulted in a mixture of 1, 2, [Cp*Fe(η5-P5)], and unidentified products according to the NMR investigations (Section 3, ESI†).
:2 molar ratio, respectively. In this case, complex 1 together with one equivalent of unreacted [Cp*Fe(η5-P5)] are obtained. It should be noted that complex 1 is not stable in solution at room temperature and further reacts slowly. However, by careful control of the reaction time, complex 1 could be isolated in a pure form (details in ESI†). In order to identify the subsequent product(s) of the degradation of complex 1, an NMR study of complex 1 was carried out. The 1H NMR spectrum showed the conversion of 1 into new species (vide infra) along with re-formation of a small amount of [Cp*Fe(η5-P5)]. However, the rate of the conversion at room temperature was found to be very slow, which prevented full conversion, even after one month (Fig. S8 and S9, ESI†). Therefore, to increase the rate, complex 1 was heated at 40 °C for 48 h (Fig. S10, ESI†). On a preparative scale, the reaction between [LGe–GeL] and [Cp*Fe(η5-P5)] was performed in toluene at 40 °C for 24 h. As a result, after work-up, a mixture of crystals was obtained, consisting mostly of 1 and [Cp*Fe(η5-P5)] along with a few brown-coloured crystals of [(LGe){(μ,η3-P5)(η1-GeL)FeCp*}] (2), which were identified by single crystal X-ray diffraction studies (for 2cf.Scheme 3 and Fig. 3). Also, attempts to obtain pure complex 2 from isolated crystalline complex 1 in toluene at 40 °C resulted in a mixture of 1, 2, [Cp*Fe(η5-P5)], and unidentified products according to the NMR investigations (Section 3, ESI†).
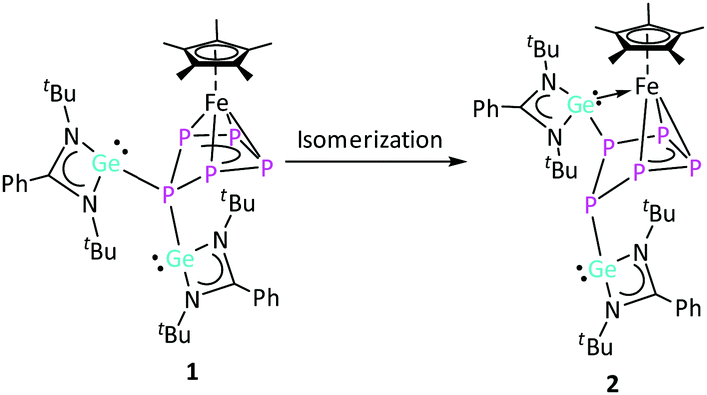 | ||
| Scheme 3 Isomerization of complex 1 to complex 2via 1,2-migration of the [LGe]+ moiety. | ||
 | ||
| Fig. 3 The molecular structure of complex 2 in the solid state. The hydrogen atoms and the solvent molecule in the unit cell are omitted for clarity. Bond lengths and angles are given in ESI.† | ||
Complex 2 is an isomer of 1, formed by an unprecedented 1,2-migration of one [LGe]+ moiety on the cyclo-P5 ring. Related 1,2-migration of a super-silyl moiety was observed during P4 activation by an olefinic silylene.6e The migrated [LGe]+ entity further inserts into one of the Fe–P bonds, resulting in an unusual η3-coordination of the cyclo-P5 ring to the [Cp*Fe]+ moiety. Despite several attempts, analytically pure complex 2 could not be isolated in meaningful yield. Nevertheless, a few crystals of complex 2 were manually separated from the mixture of crystals (1, 2 and [Cp*Fe(η5-P5)]) for NMR investigations and single crystal X-ray diffraction analysis. The Fe–P5 (2.3318(8) Å) and Fe–P4 (2.3404(7) Å) bond distances in complex 2 are similar to the Fe–P bond lengths in 1. On the other hand, the Fe–P3 (2.3934(8) Å) bond distance is significantly longer, which could be attributed to the insertion of the [LGe]+ moiety into the adjacent Fe–P2 bond. The Ge2–Fe bond length (2.2768(5) Å) is similar to the literature reported values.27 The Ge1–P1 bond length (2.4286(7) Å) in complex 2 is slightly shorter than the Ge–P distance in complex 1 (average 2.4686 Å). The distance of the newly formed Ge2–P2 bond (2.3142(7) Å) is on the shorter end of the reported Ge–P bond lengths.26,28 The P1–P2 (2.1875(10) Å), P3–P4 (2.1195(10) Å), and P4–P5 (2.1584(10) Å) bond distances are in between P–P single and double bonds. The P1–P5 (2.2153(10) Å) and P2–P3 (2.3208(11) Å) distances are consistent with a single and elongated single bond, respectively.16a,29
The 1H NMR spectrum of 2 showed a very slight downfield shift of the Cp*-methyl protons upon isomerization of 1 to 2 (δ 1.90 vs. 1.91, respectively) (Fig. S6, ESI†). In addition, four new resonances at δ = 1.10, 1.11, 1.42, and 1.52 ppm were detected in the aliphatic region, corresponding to the tBu groups of complex 2. Notably, the presence of four resonances indicates the non-equivalence of the tBu groups, which suggests a rigid conformation and restricted rotation of the [LGe] units. In agreement with the molecular structure of 2, the 31P{1H} NMR spectrum showed a set of five multiplets at δ = −43.0, −6.8, 72.4, 90.4, and 252.5 ppm, corresponding to five chemically non-equivalent P atoms. The different coupling constants were successfully assigned by iterative simulations of the spectrum (Section 4.2, ESI†).
In conclusion, we have investigated the reactivity of base-stabilized chloro-germylene and di-germylene with [Cp*Fe(η5-P5)] and [{K(dme)}2Cp*Fe(η5-P5)]. In contrast to the chloro-silylene [LSiCl], the corresponding heavier analogue, [LGeCl], did not show any reaction with [Cp*Fe(η5-P5)]. However, a stepwise reaction was observed for the di-germylene [LGe–GeL]. In the first step, the reductive homolytic cleavage of the Ge–Ge single bond in [LGe–GeL] resulted in a two-electron reduction of [Cp*Fe(η5-P5)] yielding complex 1. Complex 1 is the first example of a bis(germylene)-functionalized transition metal polyphosphide complex. Interestingly, NMR monitoring of the reaction showed that complex 1 isomerizes to a new complex, 2, even at room temperature. Complex 2 is formed by an unprecedented 1,2-migration of one [LGe]+ moiety on the cyclo-P5 ring. The formation of 1 and its subsequent isomerization leading to 2 sheds light on potential elusive intermediates that were not identified using the more reactive silicon analogues [LSiCl] and [LSi–SiL], because such intermediates were too reactive to be isolated.16a On the other hand, the reactivity observed clearly shows the difference in the reactivity of low-valent Si compounds in comparison to that of the corresponding Ge derivatives towards a metal-coordinated polyphosphide. Compounds with low-valent Si atoms were transformed into Si(IV) species by reducing cyclo-P5, however, the Ge homologues stayed at a low oxidation state, Ge(II), and displaced one P atom of the cyclo-P5 ring coordinated to the [Cp*Fe]+ moiety.
The authors are grateful to the Deutsche Forschungsgemeinschaft (DFG) (No. 266153560, Ro2008/17-2 and Sche 384/33-2).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Märkl, Angew. Chem. Int. Ed., 1965, 4, 1023 CrossRef.
- L. Bialy and H. Waldmann, Angew. Chem. Int. Ed., 2005, 44, 3814 CrossRef CAS PubMed.
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77 CrossRef CAS PubMed.
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed.
- (a) H. H. Karsch, R. Richter, A. Schier, M. Heckel, R. Ficker and W. Hiller, J. Organomet. Chem., 1995, 501, 167 CrossRef CAS; (b) E. Herdtweck, F. Jäkle and M. Wagner, Organometallics, 1997, 16, 4737 CrossRef CAS.
- (a) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (b) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (c) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (d) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169 RSC; (e) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476 RSC.
- (a) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem. Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed; (b) A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem. Int. Ed., 2005, 44, 7729 CrossRef CAS PubMed; (c) Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem. Int. Ed., 2007, 46, 4511 CrossRef CAS PubMed; (d) J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem. Int. Ed., 2009, 48, 295 CrossRef CAS PubMed; (e) K. Schwedtmann, J. Haberstroh, S. Roediger, A. Bauzá, A. Frontera, F. Hennersdorf and J. J. Weigand, Chem. Sci., 2019, 10, 6868 RSC.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180 CrossRef CAS PubMed; (b) C. L. Dorsey, B. M. Squires and T. W. Hudnall, Angew. Chem. Int. Ed., 2013, 52, 4462 CrossRef CAS PubMed.
- (a) M. Driess, A. D. Fanta, D. R. Powell and R. West, Angew. Chem. Int. Ed., 1989, 28, 1038 CrossRef; (b) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem. Int. Ed., 2011, 50, 11786 CrossRef CAS PubMed; (c) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem. Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed.
- J. W. Dube, C. M. E. Graham, C. L. B. Macdonald, Z. D. Brown, P. P. Power and P. J. Ragogna, Chem. – Eur. J., 2014, 20, 6739 CrossRef CAS PubMed.
- (a) M. Baudler, Angew. Chem. Int. Ed., 1982, 21, 492 CrossRef; (b) H. Grützmacher, Z. Anorg. Allg. Chem., 2012, 638, 1877 CrossRef.
- O. J. Scherer and T. Brück, Angew. Chem. Int. Ed., 1987, 26, 59 Search PubMed.
- (a) J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781 CrossRef CAS PubMed; (b) M. Scheer, L. J. Gregoriades, A. V. Virovets, W. Kunz, R. Neueder and I. Krossing, Angew. Chem. Int. Ed., 2006, 45, 5689 CrossRef CAS PubMed; (c) M. Scheer, A. Schindler, R. Merkle, B. P. Johnson, M. Linseis, R. Winter, C. E. Anson and A. V. Virovets, J. Am. Chem. Soc., 2007, 129, 13386 CrossRef CAS PubMed; (d) M. Scheer, Dalton Trans., 2008, 4372 RSC; (e) F. Dielmann, A. Schindler, S. Scheuermayer, J. Bai, R. Merkle, M. Zabel, A. V. Virovets, E. V. Peresypkina, G. Brunklaus, H. Eckert and M. Scheer, Chem. – Eur. J., 2012, 18, 1168 CrossRef CAS PubMed; (f) C. Schwarzmaier, A. Schindler, C. Heindl, S. Scheuermayer, E. V. Peresypkina, A. V. Virovets, M. Neumeier, R. Gschwind and M. Scheer, Angew. Chem. Int. Ed., 2013, 52, 10896 CrossRef CAS PubMed; (g) M. Fleischmann, S. Welsch, H. Krauss, M. Schmidt, M. Bodensteiner, E. V. Peresypkina, M. Sierka, C. Gröger and M. Scheer, Chem. – Eur. J., 2014, 20, 3759 CrossRef CAS PubMed.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem. Int. Ed., 2013, 52, 2972 CrossRef CAS PubMed.
- (a) T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem. Int. Ed., 2011, 50, 9491 CrossRef CAS PubMed; (b) T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183 RSC; (c) E. Mädl, M. V. Butovskii, G. Balázs, E. V. Peresypkina, A. V. Virovets, M. Seidl and M. Scheer, Angew. Chem. Int. Ed., 2014, 53, 7643 CrossRef PubMed; (d) C. Schoo, S. Bestgen, M. Schmidt, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. Commun., 2016, 52, 13217 RSC.
- (a) R. Yadav, T. Simler, S. Reichl, B. Goswami, C. Schoo, R. Köppe, M. Scheer and P. W. Roesky, J. Am. Chem. Soc., 2020, 142, 1190 CrossRef CAS PubMed; (b) R. Yadav, T. Simler, B. Goswami, C. Schoo, R. Köppe, S. Dey and P. W. Roesky, Angew. Chem. Int. Ed., 2020, 59, 9443 CrossRef CAS PubMed.
- (a) C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem. Int. Ed., 2006, 45, 3948 CrossRef CAS PubMed; (b) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed.
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311 CrossRef CAS PubMed.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem. Int. Ed., 2009, 48, 8536 CrossRef CAS PubMed.
- S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459 CrossRef CAS.
- X. Chen, T. Simler, R. Yadav, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 9315 RSC.
- Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem. Int. Ed., 2017, 56, 4333 CrossRef CAS PubMed.
- G. E. Quintero, I. Paterson-Taylor, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2016, 45, 1930 RSC.
- (a) S. Sinhababu, R. K. Siwatch, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2012, 51, 9240 CrossRef CAS PubMed; (b) Y.-L. Shan, B.-X. Leong, H.-W. Xi, R. Ganguly, Y. Li, K. H. Lim and C.-W. So, Dalton Trans., 2017, 46, 3642 RSC; (c) M. K. Sharma, D. Singh, P. Mahawar, R. Yadav and S. Nagendran, Dalton Trans., 2018, 47, 5943 RSC.
- (a) J. S. Figueroa and C. C. Cummins, Angew. Chem. Int. Ed., 2005, 44, 4592 CrossRef CAS PubMed; (b) S. Mitzinger, J. Bandemehr, K. Reiter, J. Scott McIndoe, X. Xie, F. Weigend, J. F. Corrigan and S. Dehnen, Chem. Commun., 2018, 54, 1421 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- (a) S. Karwasara, R. K. Siwatch, C. K. Jha and S. Nagendran, Organometallics, 2015, 34, 3246 CrossRef CAS; (b) N. Deak, P. M. Petrar, S. Mallet-Ladeira, L. Silaghi-Dumitrescu, G. Nemeş and D. Madec, Chem. – Eur. J., 2016, 22, 1349 CrossRef CAS PubMed.
- (a) O. T. Summerscales, M. M. Olmstead and P. P. Power, Organometallics, 2011, 30, 3468 CrossRef CAS; (b) J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem. Int. Ed., 2012, 51, 8611 CrossRef CAS PubMed; (c) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem. Int. Ed., 2012, 51, 6167 CrossRef CAS PubMed; (d) R. K. Siwatch, D. Yadav, G. Mukherjee, G. Rajaraman and S. Nagendran, Inorg. Chem., 2013, 52, 13384 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, IR spectra and X-ray crystallographic details of complexes 1 and 2. CCDC 2007825 (1) and 2007826 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03920a |
| This journal is © The Royal Society of Chemistry 2020 |