
Role of electrolyte composition on the acid stability of mixed-metal oxygen evolution catalysts†
Nancy
Li
,
Thomas P.
Keane
,
Samuel S.
Veroneau
and
Daniel G.
Nocera
 *
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. E-mail: dnocera@fas.harvard.edu
First published on 30th July 2020
Abstract
Acid stability in catalysts that promote the oxygen evolution reaction (OER) may be achieved through either the introduction of electrolyte-modulated self-healing processes, or the fixation of OER active sites within stable, conductive oxide matricies. By varying the nature of the electrolyte, pH, buffer concentration, and ionic strength, the contributions of these two effects may be deconvoluted. Furthermore, we find that the nature of the buffer is capable of engendering OER with exceptional acid stability at moderate overpotentials.
The use of sunlight to produce liquid fuels directly from CO2 is an important strategy to diversifying the global fuel supply in a renewable and sustainable manner.1–4 Viable solar-generated liquid fuel processes must proceed at appreciable rates, at high energy efficiency (low overpotential) and with high carbon atom economy. While CO2 reduction to fuels has been demonstrated at synthetically relevant rates,5 the process confronts the limitation of low carbon efficiency owing to the non-faradaic reaction of HO− with CO2 to produce super-stoichiometric amounts of carbonate, severely compromising the atom efficiency of CO2-to-fuels catalysis.6 As has recently been demonstrated, a cell equilibrium is established with the complete consumption of HO− by CO2 to produce a purely bicarbonate electrolyte.7 This acid–base chemistry results in a loss of cell voltage and, more importantly, in extremely low CO2-to-fuel carbon yields. The time for such conversion to carbonate depends on a cell's reservoir of hydroxide, giving rise to a characteristic S-curve where this hydroxide reservoir absorbs CO2 until base is depleted, after which there is a rapid rise and then levelling upon concentration of a pure carbonate electrolyte. Cells reporting high current density are typically operated for shorter times than this transition to steady-state and consequently do not reflect the actual equilibrium conditions confronting a viable solar to liquid fuels process. Thus, the CO32− problem is currently the greatest single obstacle to achieving a viable photoelectrochemical fuels synthesis.
Carbonate formation may be mitigated by performing the CO2 reduction reaction (CO2RR) in acidic solutions, thus providing an imperative for the design of acid stable catalysts for the anodic reaction (i.e., the oxygen evolution reaction, OER), which provides reducing equivalents and protons for CO2RR. We have developed OER catalysts based on cobalt8 and nickel9 that can drive OER in water (pH > 5) with rapid kinetics and good energy efficiency. In more acidic conditions (pH < 5), we have further shown that, by tuning the chemistry of self-healing,10 metallate oxygen evolving catalysts (M-OECs) may be stabilized in acidic solution.11 This stability may be enhanced further by embedding M-OECs in acid-stable metal oxide frameworks, such as MnOx or PbOx.12 Using this approach, a series of acid-stable mixed-metal OECs, including CoFePbOx, are able to maintain activity at pH 2.5 for >30 h. In such systems, the electrolyte plays an active role as a proton mediator for OER as well as a structural element in establishing the equilibrium needed for the self-healing of embedded active sites. We report herein studies seeking to explicitly define the role of electrolyte composition on film stability by comparing the OER activity of CoFePbOx in phosphate (H3PO4/KH2PO4, KPi) and sulphate (KHSO4/K2SO4, KSi), as these electrolytes possess similar chemical forms and pKas (pKa1(H3PO4) = 2.15 while pKa2(H2SO4) = 2.00).13 We find that modulating the nature, concentration and pH of the electrolyte allows us to discern the contributions of self-healing and matrix-stabilization to catalyst longevity. Moreover, as an outgrowth of these studies, we find that when operated in sulphate buffer (in the absence of phosphate), the PbOx framework can facilitate OER with exceptional acid stability at moderate overpotentials.
CoPbOx and CoFePbOx catalysts exhibit similar morphologies, as observed by SEM images of electrodeposited films, which are distinct from that of the filamentous PbOx matrix (Fig. 1). EDS mapping indicates that the component metals are distributed homogeneously throughout the catalyst films (Fig. S1, ESI†). Both films in Pi and Si perform OER with faradaic efficiencies of 100%.
 | ||
| Fig. 1 SEM images of (a) PbOx, (b) CoPbOx, (c) CoFePbOx, and (d) FePbOx for comparison. All samples were prepared on FTO substrate, and scale bar indicates 300 nm. | ||
Chronopotentiometry measurements were performed on CoFePbOx films at a constant current of 1 mA cm−2. The films were immersed in stirred solutions containing either 100 mM of KPi, 100 mM of KSi, or 100 mM each of KPi and KSi. All solutions were buffered to pH 2.15 and contained enough KNO3 supporting electrolyte to ensure a total ionic strength of 1.1 M. Representative chronopotentiometric traces are shown in Fig. 2. After a brief initial capacitive period, the operating potential for CoFePbOx films settled at ∼1.6 V vs. NHE, at which OER was sustained (0.5 V overpotential for water splitting at pH = 2.15). After a period of operation, the film potentials underwent an inflection, rising rapidly before plateauing at an overpotential of 0.7 V for KSi or 1.6 V for KPi and KPi/KSi electrolytes, indicative of OER at the PbOx matrix in KSi and FTO substrate in KPi, respectively. Direct O2 measurements for CoFePbOx films operated in KPi and KSi electrolyte solutions before and after inflection show 100% faradaic efficiency for OER, ruling out the possibility of unproductive oxidation reactions. Hence, the inflection time was used as a metric for film stability. On this basis, films operated in solutions buffered with 100 mM KPi display extended stability at lowest overpotential as compared to those buffered with 100 mM KSi.
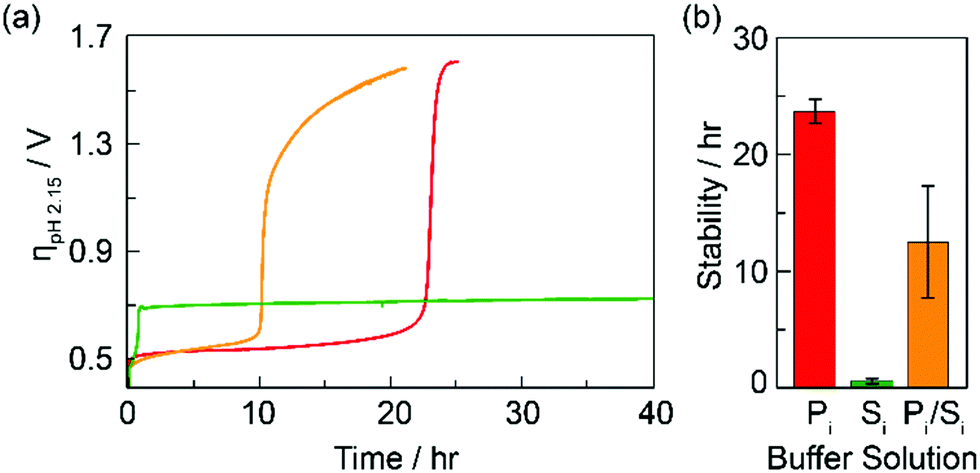 | ||
| Fig. 2 Stability of CoFePbOx films performing OER at 1 mA cm−2 in buffer solutions (pH 2.15) of 1 M KNO3 + 0.1 M KPi (red, Pi), 1 M KNO3 + 0.1 M KSi (green, Si), or 0.8 M KNO3 + 0.1 M KPi + 0.1 M KSi (orange, Pi/Si): (a) chronopotentiometry curves, and (b) bar graphs indicating average stability (time required to reach the plateau current). ηpH2.15 = Eappl − Etherm where Etherm is the thermodynamic potential for water splitting at pH = 2.15 and Eappl = applied potential. | ||
Film stability was examined for KPi and KSi electrolyte concentrations of 100 mM and 250 mM and at different pHs (Fig. S2, ESI†). For different concentrations of electrolyte, the overall buffer capacity was conserved in each electrolyte (see ESI†). At a concentration of 100 mM at pH 2.15, KPi and KSi buffers have a buffer capacity, β, of 0.074 and 0.072, respectively. At a concentration of 250 mM at pH 2.15, KPi and KSi buffers have a buffer capacity of 0.160 and 0.156, respectively. For both KPi and KSi, increasing buffer concentration led to a marginal decrease in film stability. This change cannot be attributed to ionic strength differences, as experiments performed in the presence of 1 M KNO3 showed no difference in the stability of the film in the presence of KPi whereas KSi exhibited a slight decrease as compared to experiments run in the absence of KNO3. More profound was the effect of pH on film stability. Representative chronopotentiometry curves and average film stability times at pH 1.03, 2.15 and 4.03 are shown in Fig. 3. For both electrolyte systems, film stability during OER operation decreased with decreasing pH; films operating in Si exhibited significantly more instability than films operated in Pi. For these films, a precipitous drop in stability is observed between pH 2.15 and pH 1.03, consistent with the essential role of monobasic phosphate in promoting self-healing (vide infra).
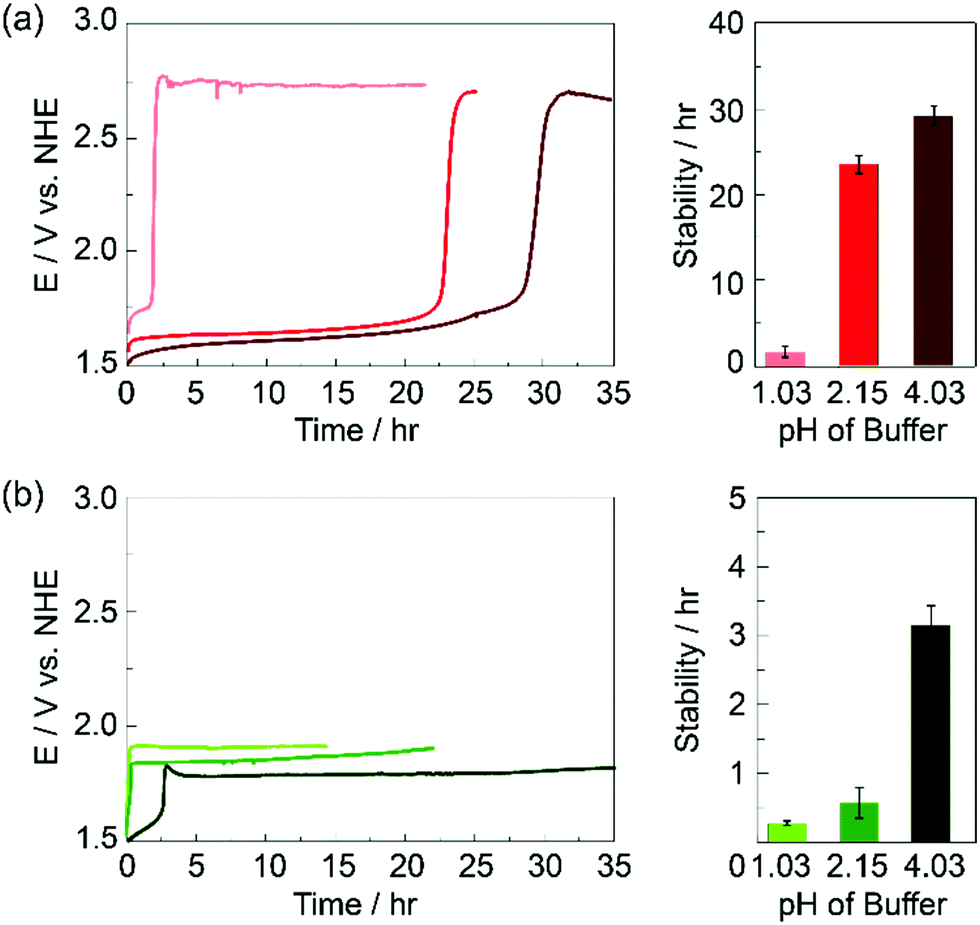 | ||
| Fig. 3 Stability of CoFePbOx films performing OER at 1 mA cm−2 in solutions containing (a) 1 M KNO3 + 0.1 M KPi and (b) 1 M KNO3 + 0.1 M KSi at different pH: 1.03 (light red and light green), pH 2.15 (red and green), and pH 4.03 (dark red and dark green). Representative chronopotentiometry curves (left), and bar graphs indicating average film stabilities (right) at each pH are shown. | ||
We compared the elemental composition of films before and after OER in different electrolyte solutions using ICP-MS (see ESI† for experimental details). The results of these experiments are summarized in Fig. 4 (Table S1, ESI†). For both KPi and KSi electrolytes, film composition does not change significantly from the as-deposited film within the first 10 min of OER. Following 40 hours of electrolysis (post-inflection for all buffer conditions) all films exhibited significant metal loss. Films operated in KPi electrolyte lose approximately 50% of their Fe content, 75% of their Co content, and 45% of their Pb content (vs. as-deposited films). This metal loss does not appear to change with addition of KNO3 or at higher concentrations of KPi. Similarly, Co loss is also observed and in even greater mangitude (88%) for CoFePbOx films operated in KSi. Interestingly, however, no Fe or Pb loss is observed for CoFePbOx films operated in KSi. The increased loss of Pb from films operated in KPi relative to KSi may reflect the formation of soluble Pb(IV) phosphate complexes, which have been reported to form electrochemically via oxidation of PbO2 in the presence of phosphoric acid.14 Formation of the corresponding sulphate complexes has not been observed.
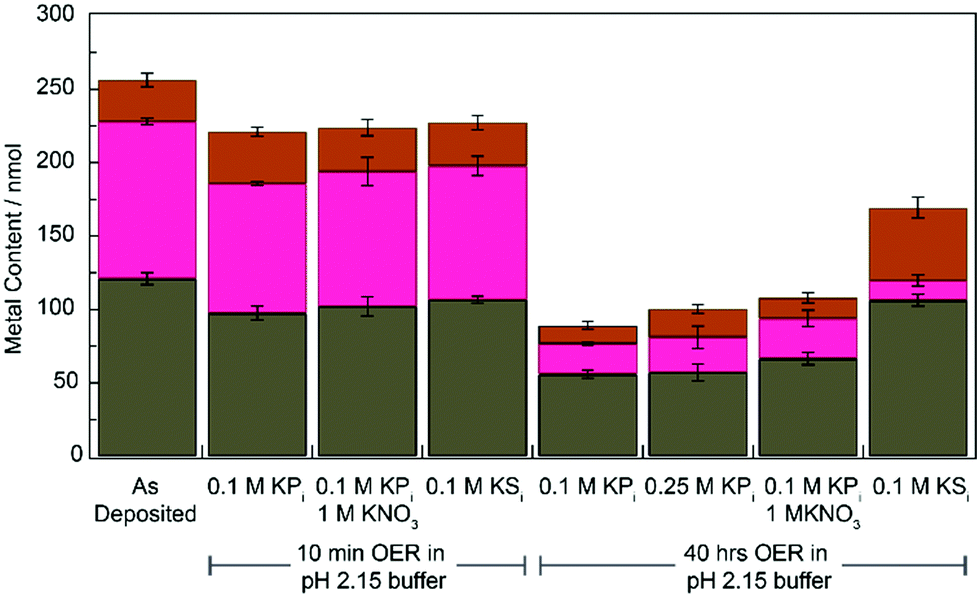 | ||
| Fig. 4 Total metal content, Pb (grey), Fe (brown), and Co (magenta) of 1 cm2 CoFePbOx films after operation in specified buffers at pH 2.15 for 0 min, 10 min, or 40 h at 1 mA cm−2 for OER. | ||
The disparate behaviour exhibited by CoFePbOx films operated in acidic KPi and KSi solutions is consistent with stabilization of the catalyst active site by (1) electrolyte-driven self-healing of active sites and (2) matrix stabilization of active sites within the PbOx framework. The work described herein now enables us to disentangle the role of self-healing and matrix stabilization in acidic media.
With regard to (1) active site stabilization via self-healing, pair distribution functional (PDF) analysis of CoPi films shows the catalyst to be composed of oxidic metallate clusters of 10–14 Co atoms.15–17 The active sites have been shown to self-heal10,18 where their self-assembly is balanced with OER such that the driving force required for self-assembly (i.e., catalyst regeneration) lies energetically within that for OER catalysis. For Co oxidic metallate clusters, when operating in acidic environments, the self-assembly process cannot be maintained owing to an inverse third-order dependence on proton activity,19 thus moving the catalyst system out of a regime of stability and imposing a limited operating lifetime on the catalyst. At pH 2.15, the dominant bases in Pi and Si are H2PO4− and SO42−, respectively, both of which are weak proton acceptors with similar basicity, and consequently neither is able to facilitate the deposition of CoPi at the same rate as HPO42−. However, PDF analysis and reaction kinetics have shown that phosphate acts as a structural element that caps the edges of the metallate clusters20 and hence provides added protection to the edge sites of the metallate clusters against protonation, thus shifting the equilibrium back towards self-assembly of the metallate active sites by impeding dissolution of the active sites during catalyst operation. This structural role of the buffer is further emphasized by the differing stability of CoFePbOx in KPi and KSi. The Pi electrolyte is known to bind oxide surfaces with higher affinity via an inner-sphere (i.e., hydrogen bonding) mechanism.21,22 Previous work has also shown that adsorbed phosphate is capable of stabilizing various metal oxide surfaces against corrosion.23,24 In contrast, Si has a significantly lower affinity for oxides, and studies have found evidence for an outer-sphere (i.e., ionic) mode of adsorption by sulphate onto these surfaces.25–27
With regard to (2) matrix stabilization, the comparison of CoPi and CoFePbOx in Fig. 5 demonstrates the effect of the lead oxide matrix in enhancing acid stability. Elemental composition data summarized in Fig. 4 reveal sustained resistance of the PbOx matrix to corrosion, further reinforcing the role of PbOx as a stable structural component.
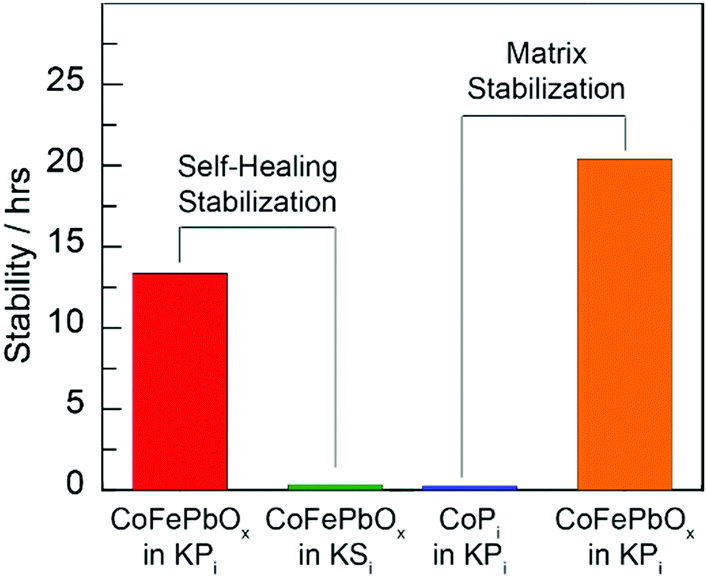 | ||
| Fig. 5 Stability ascribed to (left) self-healing by comparison of CoFePbOx films operating at 1 mA cm−2 in 1 M KNO3 and either 0.25 M KPi (red) or 0.25 M KSi (green) solution, buffered to pH 2.15, and (right) matrix stabilization by comparison of CoPi (blue) and CoPbOx (orange) films operating at 1 mA cm−2 in a 1 M KNO3, 0.25 M KPi, solution, buffered to pH 2.5 (ref. 12). | ||
The chronopotentiometric trace in Fig. 2a for CoFePOx in Si is intriguing, and highlights the important role that electrolyte can have on OER film stability and activity. The trace begins at an overpotential similar to that observed for CoFePbOx in Pi. After approximately 1 h, the operating potential rises, consistent with the loss of Co from the film (Fig. 4) and the trace stabilizes at an overpotential of 0.7 V, which is almost 1 V lower than that observed for films operated in KPi following similar Co loss. Control experiments indicate that this potential is representative of OER on a PbOx film when operated in KSi buffer (Fig. S3, ESI†). When PbOx is operated at a similar pH, but in the presence of KPi, a 1.7 V overpotential is required to achieve the same current density.12 Though PbOx in KSi requires a higher overpotential for OER than CoFePbOx, it is exceedingly stable in acid, maintaining activity for over 150 h with no sign of degradation (Fig. S4, ESI†). The stark contrast in operating potentials for PbOx in KPiversus KSi may result from differences in oxide surface-binding between phosphate and sulphate, as described above. Notably, a similar explanation has been proposed in the lead-acid battery literature where phosphoric acid is known to be an effective additive for suppressing OER.28 These results demonstrate the impact that buffer identity can have on the operation and activity of OER catalysts in acid and forecasts the need to consider the role of electrolyte in the development of active, stable, and earth-abundant OER catalysts.
Material is based upon work supported under the Solar Photochemistry Program of the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences of the U. S. Department of Energy DE-SC0017619. T. P. K. acknowledges support from a Graduate Research Fellowship from the National Science Foundation. We thank Dr Matthew Nava for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- D. Abbott, Proc. IEEE, 2010, 98, 42–66 CAS.
- F. P. G. de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- B. A. Zhang, C. Costentin and D. G. Nocera, Joule, 2019, 3, 1565–1568 CrossRef.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. Dinca, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef CAS PubMed.
- C. Costentin and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13380–13384 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2018, 10, 24–30 CrossRef CAS PubMed.
- R. F. Amlie and T. A. Berger, J. Electroanal. Chem. Interfacial Electrochem., 1972, 36, 427–433 CrossRef CAS.
- C. L. Farrow, D. K. Bediako, Y. Surendranath, D. G. Nocera and S. J. L. Billinge, J. Am. Chem. Soc., 2013, 135, 6403–6406 CrossRef CAS PubMed.
- C. N. Brodsky, D. K. Bediako, C. Shi, T. P. Keane, C. Costentin, S. J. L. Billinge and D. G. Nocera, ACS Appl. Energy Mater., 2019, 2, 3–12 CrossRef CAS.
- P. Du, O. Kokhan, K. W. Chapman, P. J. Chupas and D. M. Tiede, J. Am. Chem. Soc., 2012, 134, 11096–11099 CrossRef CAS PubMed.
- D. K. Bediako, A. M. Ullman and D. G. Nocera, Catalytic Oxygen Evolution by Cobalt Oxido Thin Films. in Topics in Current Chemistry, ed. H. Tuysuz and C. K. Chan, Springer Publishing AG, Switzerland, 2016, 173–213 Search PubMed.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326–6336 CrossRef CAS PubMed.
- G. Kwon, H. Jang, J. S. Lee, A. Mane, D. J. Mandia, S. R. Soltau, L. M. Utschig, A. B. F. Martinson, D. M. Tiede, H. Kim and J. Kim, J. Am. Chem. Soc., 2018, 140, 10710–10720 CrossRef PubMed.
- Y. Arai and D. L. Sparks, J. Colloid Interface Sci., 2001, 241, 317–326 CrossRef CAS.
- E. Bae and W. Choi, J. Phys. Chem. B, 2006, 110, 14792–14799 CrossRef CAS PubMed.
- E. D. Laudadio, J. W. Bennett, C. M. Green, S. E. Mason and R. J. Hamers, Environ. Sci. Technol., 2018, 52, 10186–10195 CrossRef CAS PubMed.
- W. Stumm and R. Wollast, Rev. Geophys., 1990, 28, 53 CrossRef.
- J. S. Geelhoed, T. Hiemstra and W. H. Van Riemsdijk, Geochim. Cosmochim. Acta, 1997, 61, 2389–2396 CrossRef CAS.
- D. D. Hansmann and M. A. Anderson, Environ. Sci. Technol., 1985, 19, 544–551 CrossRef CAS PubMed.
- L. Charlet, N. Dise and W. Stumm, Agric., Ecosyst. Environ., 1993, 47, 87–102 CrossRef CAS.
- M. P. Vinod, K. Vijayamohanan and S. N. Joshi, J. Power Sources, 1998, 70, 103–105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional tables and figures. See DOI: 10.1039/d0cc03863f |
| This journal is © The Royal Society of Chemistry 2020 |