
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrodeoxygenation of hydroxyacetophenones to ethyl-substituted phenol derivatives using a FeRu@SILP catalyst†
Lisa
Goclik
ab,
Lisa
Offner-Marko
ab,
Alexis
Bordet
 *a and
Walter
Leitner
*ab
*a and
Walter
Leitner
*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: alexis.bordet@cec.mpg.de; walter.leitner@cec.mpg.de
bInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 16th July 2020
Abstract
The selective hydrodeoxygenation of hydroxyacetophenone derivatives is achieved opening a versatile pathway for the production of valuable substituted ethylphenols from readily available substrates. Bimetallic iron ruthenium nanoparticles immobilized on an imidazolium-based supported ionic liquid phase (Fe25Ru75@SILP) show high activity and stability for a broad range of substrates without acidic co-catalysts.
Although substituted ethylphenols have attracted considerable attention due to their importance as building blocks for the production of fine chemicals,1 polymers,2 and pharmaceuticals,3 their synthesis remains particularly challenging. The Friedel–Crafts alkylation of phenols4 to produce ethylphenol derivatives is indeed difficult and requires harsh conditions, since the –OH group of phenol coordinates with classical Lewis acidic catalysts and prevents the reaction. In addition, this pathway offers poor chemo- and regioselectivity as well as a very limited substrate scope. Several alternative methods providing access to ethylphenol derivatives have been developed, including in particular the hydrogenolysis of diarylethers and lignin model substrates,5 and the hydrogenation of vinylphenol derivatives.6 These approaches suffer from significant limitations, however, such as low catalyst activity,5b,c,6a low selectivity,5a,e,f low stability/recyclability,5a,d,f or limited substrate scope.5c–e The high costs of vinylphenols are prohibitive for their use as starting materials in most commercial applications.6 In this context, the hydrodeoxygenation of easily accessible hydroxyacetophenone derivatives – obtained for example through Friedel–Crafts acylation,7 Fries rearrangement8 or oxidative depolymerization of lignin9 – could offer an attractive alternative. Robust synthetic methods based on these transformations require the design of catalytic systems capable of hydrogenating and deoxygenating aromatic ketones, while leaving the aromaticity of the ring untouched (Fig. 1A). Recent efforts resulted in the development of catalysts with promising performance for the selective hydrogenation and hydrodeoxygenation of aromatic substrates.10,11 In particular, we introduced a bifunctional catalyst composed of bimetallic iron–ruthenium nanoparticles immobilized on an acid-functionalized supported ionic liquid phase (Fe25Ru75@SILP + IL-SO3H) and demonstrated its excellent activity, selectivity, and stability for the hydrodeoxygenation of benzylic and non-benzylic ketones.10a However, despite its versatility, this bifunctional catalyst cannot be employed to convert hydroxyacetophenone derivatives due to competing acid-catalyzed side-reactions.12 Now we report that we were able to close this gap in the substrate scope by utilizing bimetallic Fe25Ru75 nanoparticles immobilized on a non-acidic imidazolium-based SILP support. The mesomeric effects activating the ketone and stabilizing the intermediate carbocation were found to be the key to high hydrodeoxygenation activity under the optimized acid-free conditions. Thus, a broad range of hydroxyacetophenone derivatives were effectively and selectively hydrodeoxygenated using the Fe25Ru75@SILP catalyst, giving access to a wide range of ethylphenols, which are very difficult to obtain through other synthetic methods. This opens a new retrosynthetic pathway for the production of valuable substituted ethylphenols from widely accessible substrates (Fig. 1).
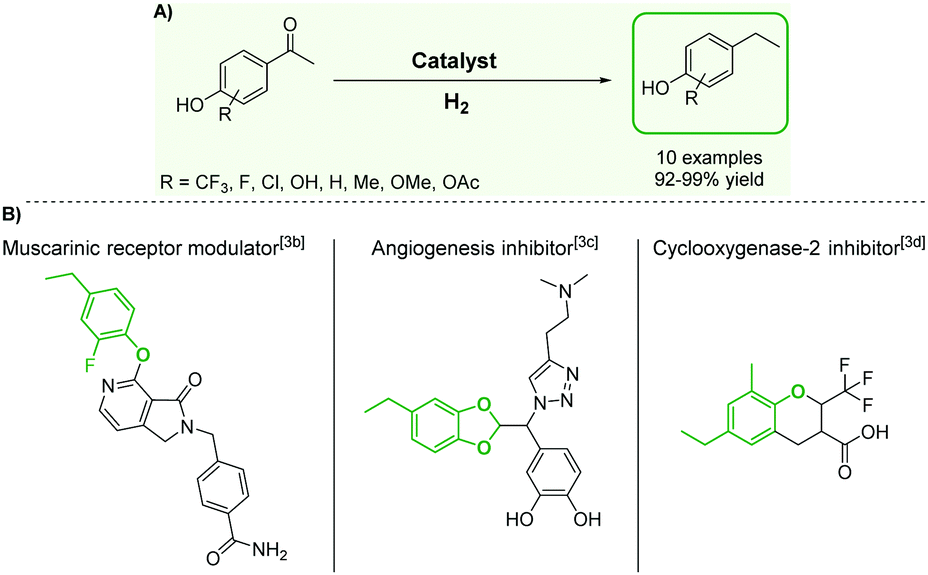 | ||
| Fig. 1 (A) Pathway for the synthesis of alkyl phenol derivatives through hydrodeoxygenation of hydroxyacetophenones. (B) Examples of applications for substituted alkyl phenols in pharmaceutical products. | ||
The Fe25Ru75@SILP catalyst (Fig. 2a) was synthesized through an organometallic approach following a recently reported procedure (see ESI† for details).10b The use of the imidazolium-based SILP is crucial for the formation of small, well-dispersed and homogeneously alloyed Fe25Ru75 NPs under these conditions, as well as for their efficient stabilization through electrosteric interactions. TEM analysis evidenced the formation of small and well-dispersed nanoparticles with an average size of 3.3 nm (Fig. 2b). The catalyst was further characterized by BET measurements, giving a surface area of 170 m2 g−1, corresponding to about 50% of the undecorated silica. SEM-EDX analysis indicated a metal loading of 0.4 mmol g−1 and a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru ratio of 25:75 (±5%), well in agreement with the theoretical values (Table S1, ESI†). The oxidation state and alloy extent of the Fe25Ru75 NPs in Fe25Ru75@SILP were investigated by XANES and EXAFS already in a previous study, evidencing zerovalent Fe and Ru atoms organized in a slightly homophilic bimetallic structure.10b
:Ru ratio of 25:75 (±5%), well in agreement with the theoretical values (Table S1, ESI†). The oxidation state and alloy extent of the Fe25Ru75 NPs in Fe25Ru75@SILP were investigated by XANES and EXAFS already in a previous study, evidencing zerovalent Fe and Ru atoms organized in a slightly homophilic bimetallic structure.10b
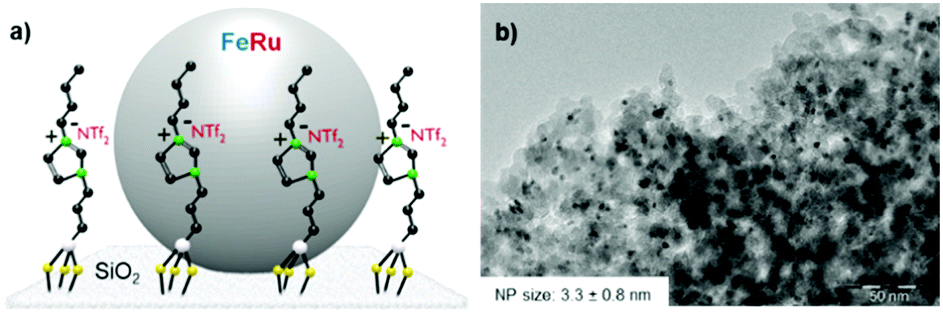 | ||
| Fig. 2 Fe25Ru75@SILP; (a) illustration of the catalytic system; (b) TEM image. | ||
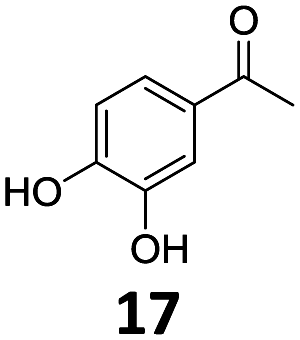
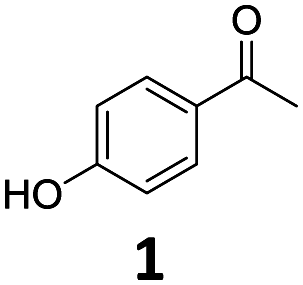
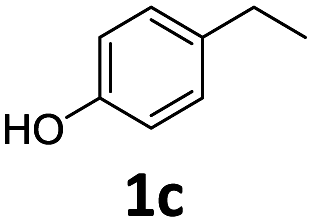
The catalytic properties of Fe25Ru75@SILP and several reference catalysts were investigated using 4-hydroxyacetophenone (1) as model substrate (Table 1). Monometallic Fe@SILP presented no activity under these conditions. Ru@SILP lead to the full but unselective conversion of the substrate, with the production of a mixture of 4-(1-hydroxy-ethyl)cyclohexan-1-ol (1g, 41%), 4′-ethylcyclohexanol (1h, 50%), and dimers (9%). As expected, the aromaticity of the ring was not conserved. The use of the recently reported Fe25Ru75@SILP+IL-SO3H catalyst10a resulted in low selectivity and low yield of the desired product (1c, 53%). Other products observed include phenol (1d, 30%), 2′4′-ethylphenol (1e, 12%) and 2′-ethylphenol (1f, 5%). The formation of these compounds is due to several acid-catalysed side-reactions such as protiodeacylation,12 Friedel–Crafts acylation and hydrodeoxygenation. In contrast, the hydrodeoxygenated product 1c was yielded quantitatively using Fe25Ru75@SILP. The hydrogenation selectivity observed here is in agreement with our previous studies showing that alloying Fe and Ru in this specific 25:75 ratio generates synergistic effects leading to a significant enhancement of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation rate as compared to monometallic Ru, while the aromatic hydrogenation is prevented.10a,b To investigate the versatility of Fe25Ru75@SILP as a hydrodeoxygenation catalyst, its activity was tested for various acetophenone derivatives under standard conditions (Table S2, ESI†) Interestingly, significant activity was observed only for acetophenone derivatives possessing functional groups providing mesomeric stabilization (–OH, –OMe, –OPh, –NH2, etc.), suggesting the importance of mesomeric effects on the hydrodeoxygenation. For the substrates, which cannot provide this stabilisation, the hydrogenation products (2a, 3a, 5a, 6a) were mostly observed. To further study the importance of the mesomeric stabilisation, 2′- and 3′-hydroxyacetophenone were considered as substrates (Table S3, ESI†). With the hydroxyl group located in ortho- (13) or para- (1) positions, the conversion, selectivity and yield of the target products are over 99%. In the case of the meta-substituted substrate 3′-hydroxyacetophenone (12), the conversion is significantly lower with 39%. The selectivity remains however excellent (95%). The results confirm that mesomeric effects play a key role activating the substrates and stabilizing the intermediate carbocation. Despite a lower reactivity, the excellent selectivity observed with 3′-hydroxyacetophenone (12) as substrate suggests that under optimized reaction conditions Fe25Ru75@SILP could effectively produce meta-ethyl phenols, which are extremely difficult to obtain through other pathways.
O hydrogenation rate as compared to monometallic Ru, while the aromatic hydrogenation is prevented.10a,b To investigate the versatility of Fe25Ru75@SILP as a hydrodeoxygenation catalyst, its activity was tested for various acetophenone derivatives under standard conditions (Table S2, ESI†) Interestingly, significant activity was observed only for acetophenone derivatives possessing functional groups providing mesomeric stabilization (–OH, –OMe, –OPh, –NH2, etc.), suggesting the importance of mesomeric effects on the hydrodeoxygenation. For the substrates, which cannot provide this stabilisation, the hydrogenation products (2a, 3a, 5a, 6a) were mostly observed. To further study the importance of the mesomeric stabilisation, 2′- and 3′-hydroxyacetophenone were considered as substrates (Table S3, ESI†). With the hydroxyl group located in ortho- (13) or para- (1) positions, the conversion, selectivity and yield of the target products are over 99%. In the case of the meta-substituted substrate 3′-hydroxyacetophenone (12), the conversion is significantly lower with 39%. The selectivity remains however excellent (95%). The results confirm that mesomeric effects play a key role activating the substrates and stabilizing the intermediate carbocation. Despite a lower reactivity, the excellent selectivity observed with 3′-hydroxyacetophenone (12) as substrate suggests that under optimized reaction conditions Fe25Ru75@SILP could effectively produce meta-ethyl phenols, which are extremely difficult to obtain through other pathways.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Xa [%] | Y1ca [%] | Y1d–fa [%] | Y1g,ha [%] |
| Reaction conditions: catalyst (37.5 mg, metal content: 0.015 mmol), substrate (0.375 mmol, 25 eq.), solvent (0.5 mL), 175 °C, H2 (50 bar), 16 h, 500 rpm.a Determined by GC-FID using tetradecane as an internal standard.b Products are 1g (41%), 1h (50%), dimers (9%).c Other products: 1d (30%), 1e (12%) and 1f (5%). | |||||
| 1 | Fe100@SILP | 0 | 0 | 0 | 0 |
| 2 | Ru100@SILP | >99 | 0 | 0 | 91b |
| 3 | Fe25Ru75@ | >99 | 53 | 47c | 0 |
| SILP+IL-SO3H | |||||
| 4 | Fe25Ru75@SILP | >99 | >99 | 0 | 0 |
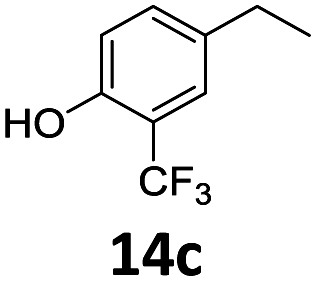
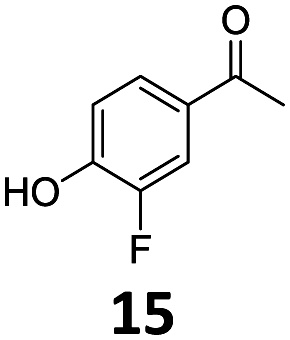
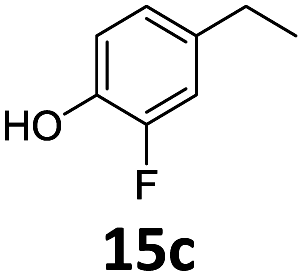
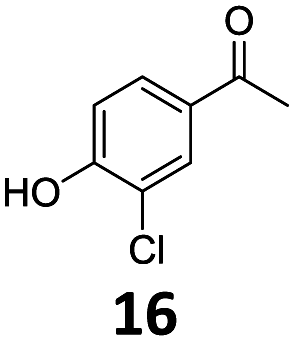
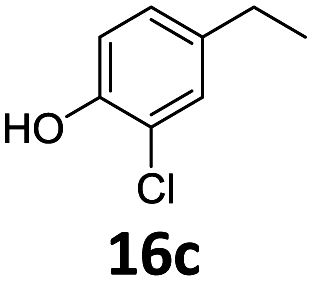
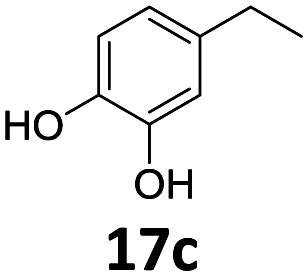
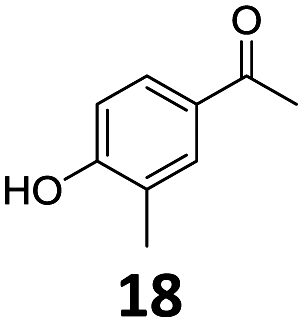
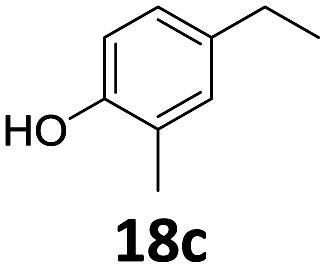
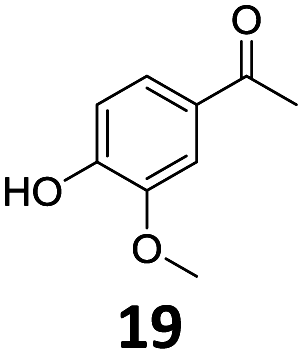
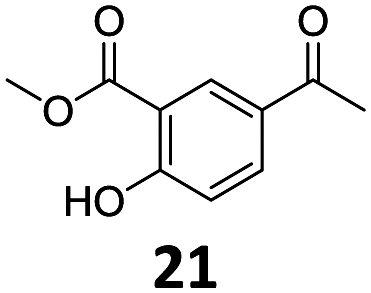
Based on these promising results, the Fe25Ru75@SILP catalyst was applied to a scope of hydroxyacetophenone derivatives with a wide range of substituent patterns (Table 2). All the substrates considered were effectively hydrodeoxygenated under these conditions, producing substituted ethylphenols in excellent selectivity and yields. Interestingly, the products 15c, 17c and 18c are building blocks used directly in the synthesis of the pharmaceutical compounds highlighted in Fig. 1B.3b–d Substrates 19 and 20 can be obtained from lignin,13 evidencing the capacity of Fe25Ru75@SILP to convert biomass-derived substrates to value added chemicals. Furthermore, the synthesis of products 14c and 15c was, to the best of our knowledge, not reported before, and this is thus the first synthetic method providing these compounds in high yields and selectivity. The production of 22c in excellent yield indicates that the length of the chain does not affect the reactivity. This opens the way to the synthesis of long chain alkyl phenols, which are key reagents for the production of alkylphenol ethoxylates, a very important class of non-ionic surfactants.14 Comparing the prices of the substrates and products at classical providers shows that in the vast majority the cases, the ethylphenol derivatives are much more expensive than the starting hydroxyacetophenones, confirming the value-adding potential through this pathway (Table S4, ESI†). The results show that the conversion of hydroxyacetophenone derivatives possessing electron-donating substituents is significantly faster than for those possessing electron-withdrawing functionalities. This is in agreement with previous observations.11d The corresponding alcohols and olefins were observed in the product mixtures at these shortened reaction times, supporting a hydrogenation/dehydration/hydrogenation sequence as plausible reaction pathway. The nature and proportion of the intermediates observed during the reactions strongly depends on the nature of the substituents, albeit no clear trend could be identified regarding the electron donating/withdrawing properties of the substituents. This most likely reflects different effects of the substituents on the rates of the individual steps of the sequence. To get further insight into the hydrodeoxygenation activity of Fe25Ru75@SILP, a time profile was recorded using 4′-hydroxyacetophenone (1) as model substrate (Fig. S1, ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Xa [%] | Sa [%] | Y*a [%] |
| Reaction conditions: catalyst (37.5 mg, metal content: 0.015 mmol), substrate (0.375 mmol, 25 eq.), solvent (0.5 mL), 175 °C, H2 (50 bar), 16 h, 500 rpm.a Determined by GC-FID using tetradecane as internal standard. *Isolated yield in parentheses.b By-products is 4-ethylphenol (4%).c By-product is methyl 2-hydroxy-5-(1-hydroxyethyl)benzoate (4%). X = conversion, S = selectivity, Y = yield. |
|||||
| 1 |
|
|
97 | >99 | 97 |
| 2 |
|
|
>99 | 96b | 96 (81) |
| 3 |
|
|
>99 | 96b | 96 |
| 4 |
|
|
>99 | >99 | >99 |
| 5 |
|
|
>99 | >99 | >99 (88) |
| 6 |
|
|
>99 | >99 | >99 (85) |
| 7 |
|
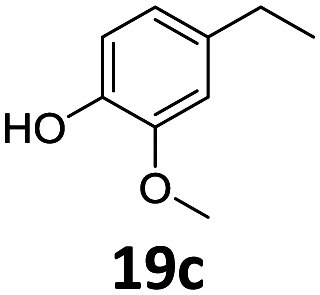
|
>99 | >99 | >99 (91) |
| 8 |
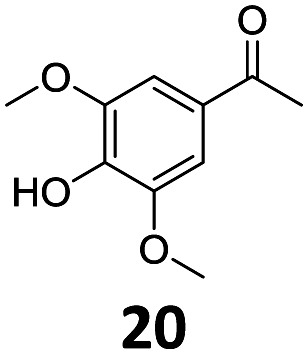
|
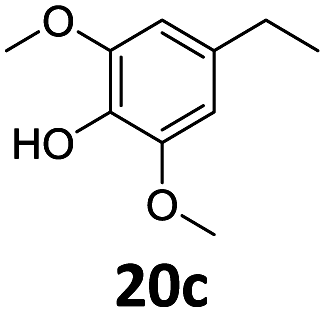
|
>99 | >99 | >99 (98) |
| 9 |
|
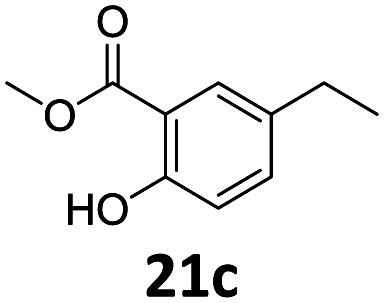
|
96 | 96c | 92 |
| 10 |
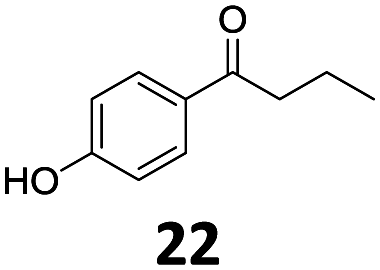
|
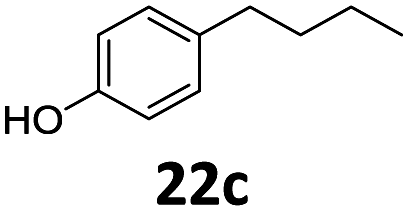
|
>99 | >99 | >99 (91) |
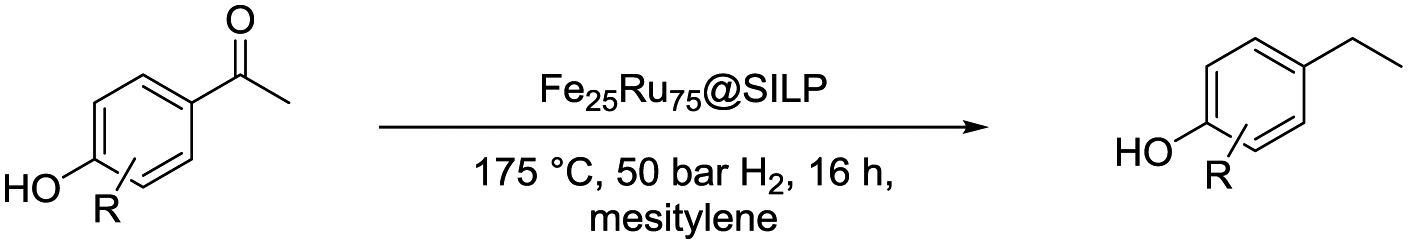
The reaction temperature was reduced to 120 °C in order to slow down the reaction and allow the observation of the intermediates. Under these conditions, the substrate was fully converted after 3 h, giving 1c in quantitative yield. 4-Vinylphenol (1b) was the only intermediate observed, suggesting a very fast deoxygenation of the alcohol intermediate 1a. Prolonging the reaction time to 16 h did not change the product distribution, highlighting the excellent selectivity of Fe25Ru75@SILP. Interestingly, the conversion of the substrate (and formation of 1c) appears to become faster with time, indicating that the reaction rate increases when the concentration of the substrates decreases. This presumably reflects the influence of the release of water during the reaction, rather than a true negative kinetic order with regards to the substrate. Mesitylene is a hydrophobic solvent, and the water released during hydrodeoxygenation accumulates on the catalyst, increasing progressively the polarity of the medium around the NPs. Consequently, the affinity of the polar substrate 1 for adsorption at the Fe25Ru75@SILP material also increases, thus resulting in an increase of the local substrate concentration around NPs and an acceleration of the reaction, even though the global substrate concentration decreases. The accumulation of water on the catalyst during the reaction was confirmed by IR spectroscopy, supporting this assumption (Fig. S2, ESI†). Similar increases in reaction rate due to high local concentrations of substrates has previously been reported for SCILL-type catalysts where the substrates are more soluble in the IL layer than in the solvent used.15 Recording a time profile in the presence of deliberately added small amounts of H2O resulted indeed in much higher initial activity, strongly supporting this hypothesis (Fig. S3, ESI†). The release of water as a co-product of the reaction is thus beneficial in this case, since it counterbalances the global decrease of the substrate concentration with time by increasing its affinity (and thus local concentration) around the active sites. This behaviour is in contrast to most of the deoxygenation catalysts previously reported, which tend to lose their activity due to the accumulation of water.5a,16 The possibility to recycle the Fe25Ru75@SILP catalyst was studied using 4′-hydroxyacetophenone (1) as substrate. Fully constant conversion and selectivity towards the formation of 4-ethylphenol (1c) were obtained in five consecutive runs without any make-up or regeneration (Fig. S4, ESI†). After 5 cycles, the catalyst was characterized by BET, TEM and SEM-EDX (Table S1, ESI†). BET analysis indicated that the textural properties of the catalyst remained largely unaffected. According to TEM analysis, the size of the NPs and their dispersion on the support did not change significantly. In addition, SEM-EDX did not evidence any significant leaching of the metal or any modification of the Fe:Ru ratio under these conditions. These results evidence the excellent stabilization of the bimetallic NPs by the SILP.
In conclusion, the organometallic approach to graft bimetallic Fe25Ru75 nanoparticles on imidazolium-modified silica materials produces a Fe25Ru75@SILP catalyst with excellent properties for the hydrodeoxygenation of hydroxyacetophenone derivatives. The presence of the OH-substituent allows mesomeric stabilization of the carbocation intermediates and is essential to observe high hydrodeoxygenation activity under these non-acidic conditions. Achieving the hydrodeoxygenation in the absence of acidic co-catalysts is beneficial for this particular class of substrates, since it prevents side-reactions such as protiodeacylation. Synthesized through a molecular approach, the Fe25Ru75@SILP catalyst was found to be highly active and stable for the selective conversion of a wide range of hydroxyacetophenones with various functional groups, opening a versatile route for the production of value-added substituted ethylphenols from widely available substrates. Preliminary results indicate that the same retrosynthetic design is applicable to ethylaniline derivatives and long chain alkyl phenols, which are key intermediates in the synthesis of pharmaceuticals and non-ionic surfactants, respectively.
The authors acknowledge financial support by the Max Planck Society and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832. Furthermore, the authors thank Alina Jakubowski, Annika Gurowski, Justus Werkmeister and Norbert Pfänder (MPI-CEC, Mülheim/Ruhr) for their support with the analytics. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.References
- R. O. Hutchins, M. K. Hutchins and I. Fleming, Comprehensive Organic Synthesis, Elsevier, Oxford, 8th edn, 1991, p. 327 Search PubMed.
- (a) D. Pan, L. Zhang, F. Li, Y. Chen, N. Tao, G. Zhang and Y. Dai, CN102719820, Faming Zhuanli Shenqing, 2012; (b) Y. Nakai and N. Haruyama, WO2019130722, 2019.
- (a) J. F. Lorenc, G. Lambeth and W. Scheffer, Alkylphenols, John Wiley & Sons, Inc., 2003 Search PubMed; (b) J. L. Wang, K. Aston, D. Limburg, C. Ludwig, A. E. Hallinan, F. Koszyk, B. Hamper, D. Brown, M. Graneto and J. T. John, Bioorg. Med. Chem. Lett., 2010, 20(23), 7164 CrossRef CAS PubMed; (c) S. Yu, J. Oh, F. Li, Y. Kwon, H. Cho, J. Shin, S. K. Lee and S. Kim, ACS Med. Chem. Lett., 2017, 8(10), 1066 CrossRef CAS PubMed; (d) M. V. Galkin, S. Sawadjoon, V. Rohde, M. Dawange and J. S. M. Samec, ChemCatChem, 2014, 6, 179 CrossRef CAS.
- G. A. Olah, Friedel–Crafts Chemistry, Wiley-VCH, NY, 1973 Search PubMed.
- (a) S. Rengshausen, F. Etscheidt, J. Großkurth, K. L. Luska, A. Bordet and W. Leitner, Synlett, 2019, 405 CAS; (b) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed; (c) F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem. Int. Ed., 2016, 55, 1474 CrossRef CAS PubMed; (d) J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554 CrossRef CAS PubMed; (e) S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef PubMed; (f) S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748 RSC.
- (a) Y. Arakawa, K. Minagawa and Y. Imada, Polym. J., 2018, 50, 941 CrossRef CAS; (b) T. Takahashi, M. Yoshimura, H. Suzuka, T. Maegawa, Y. Sawama, Y. Monguchi and H. Sajiki, Tetrahedron, 2012, 68, 8293 CrossRef CAS.
- (a) H. Heaney, B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, Pergamon Press, Oxford, UK, 1991, vol. 2, p. 733 Search PubMed; (b) R. Faust, US Pat., US 20170001936, 2017, p. 20 Search PubMed.
- (a) R. W. Stoughton, R. Baltzly and A. Bass, J. Am. Chem. Soc., 1934, 56, 2007 CrossRef CAS; (b) I. Jeon and I. K. Mangion, Synlett, 2012, 1927 CrossRef CAS; (c) H. Paghandeh, H. Saeidian and M. Ghaffarzadeh, Lett. Org. Chem., 2018, 15, 809–814 CrossRef CAS.
- (a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed; (b) Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614 CrossRef CAS PubMed.
- (a) L. Offner-Marko, A. Bordet, G. Moos, S. Tricard, S. Rengshausen, B. Chaudret, K. L. Luska and W. Leitner, Angew. Chem. Int. Ed., 2018, 57, 12721–12726 CrossRef CAS PubMed; (b) K. L. Luska, A. Bordet, S. Tricard, I. Sinev, W. Grünert, B. Chaudret and W. Leitner, ACS Catal., 2016, 6, 3719 CrossRef CAS; (c) S. El Sayed, A. Bordet, C. Weidenthaler, W. Hetaba, K. L. Luska and W. Leitner, ACS Catal., 2020, 10, 2124 CrossRef CAS; (d) G. Moos, M. Eimondts, A. Bordet and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 11977–11983 CrossRef CAS PubMed.
- (a) H. Wang, L. Li, X.-F. Bai, J.-Y. Shang, K.-F. Yang and L.-W. Xua, Adv. Synth. Catal., 2013, 355, 341 CAS; (b) L. Petitjean, R. Gagne, E. S. Beach, D. Xiao and P. T. Anastas, Green Chem., 2016, 18, 150 RSC; (c) D. Verma, R. Insyani, H. S. Cahyadi, J. Park, S. M. Kim, J. M. Cho, J. W. Bae and J. Kim, Green Chem., 2018, 20, 3253 RSC; (d) J. M. Asensio, A. B. Miguel, P.-F. Fazzini, P. W. N. M. van Leeuwen and B. Chaudret, Angew. Chem., 2019, 131, 11428 Search PubMed.
- M. Koželj and A. Petrič, Synlett, 2007, 1699 Search PubMed.
- (a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559 CrossRef CAS PubMed; (b) J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554 CrossRef CAS PubMed; (c) A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 7526 CrossRef PubMed.
- (a) R. S. Bolva and M. K. Markoc, Tenside, Surfactants, Deterg., 1981, 18, 37 Search PubMed; (b) H.-D. Dörfler, Interfacial and Colloid Chemistry (in German), VCH Verlagsges, Weinheim, Germany, 1994 Search PubMed.
- (a) M. Haumann and P. Wasserscheid, SILP and SCILL Catalysis, Royal Society of Chemistry, 2014 Search PubMed; (b) M. Lijewski, J. M. Hogg, M. Swadzba-Kwasny, P. Wasserscheid and M. Haumann, RSC Adv., 2017, 7, 27558 RSC.
- (a) K. L. Luska, P. Migowski, S. El Sayed and W. Leitner, Angew. Chem. Int. Ed., 2015, 54, 14750 CrossRef PubMed; (b) Y. Liu, M. A. Mellmer, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2015, 8, 3983 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc03695a |
| This journal is © The Royal Society of Chemistry 2020 |