
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural evidence for a reaction intermediate mimic in the active site of a sulfite dehydrogenase†
Ahmed
Djeghader
 a,
Melanie
Rossotti
b,
Saleh
Abdulkarim‡
a,
Frédéric
Biaso
b,
Guillaume
Gerbaud
b,
Wolfgang
Nitschke
b,
Barbara
Schoepp-Cothenet
*b,
Tewfik
Soulimane
*a and
Stéphane
Grimaldi
*b
a,
Melanie
Rossotti
b,
Saleh
Abdulkarim‡
a,
Frédéric
Biaso
b,
Guillaume
Gerbaud
b,
Wolfgang
Nitschke
b,
Barbara
Schoepp-Cothenet
*b,
Tewfik
Soulimane
*a and
Stéphane
Grimaldi
*b
aDepartment of Chemical Sciences and Bernal Institute, University of Limerick, Ireland. E-mail: Tewfik.soulimane@ul.ie
bAix Marseille Univ, CNRS, BIP UMR7281, Marseille, France. E-mail: schoepp@imm.cnrs.fr; stephane.grimaldi@univ-amu.fr
First published on 21st July 2020
Abstract
By combining X-ray crystallography, electron paramagnetic resonance techniques and density functional theory-based modelling, we provide evidence for a direct coordination of the product analogue, phosphate, to the molybdenum active site of a sulfite dehydrogenase. This interaction is mimicking the still experimentally uncharacterized reaction intermediate proposed to arise during the catalytic cycle of this class of enzymes. This work opens new perspectives for further deciphering the reaction mechanism of this nearly ubiquitous class of oxidoreductases.
Eukaryotic sulfite oxidases (SOs) and homologous prokaryotic sulfite dehydrogenases (SDHs) are mononuclear molybdoenzymes that catalyze sulfite oxidation by direct oxygen atom transfer from water to the substrate: SO32− + H2O → SO42− + 2H+ + 2e−.1 They play a key role in sulfur detoxification, notably in the final step of the degradation pathway of sulfur-containing amino acids. In humans, SO deficiency is a genetic disease that causes severe neonatal neurological problems and often early death.2 In bacteria, SDHs have a central role in converting SO32− formed during dissimilatory oxidation of reduced sulfur compounds, and are generally linked to energy conservation.3 In contrast to SOs, SDHs are unable to use O2 as a direct electron acceptor. SO32− oxidation in these Sulfite Oxidizing Enzymes (SOEs) occurs at their conserved Mo center which cycles between the Mo(IV), Mo(V) and Mo(VI) states (Fig. S1, ESI†).4 The combined use of X-ray crystallography, extended X-ray absorption fine structure (EXAFS), various electron paramagnetic resonance (EPR) spectroscopic methods and theoretical modelling has been crucial to resolve the geometrical and electronic structure of the Mo cofactor (Moco) at the active site of SOEs.1,5–7 The metal ion is coordinated to five ligand donor atoms in a pseudo-square pyramidal geometric arrangement: three equatorial S donor atoms (one from a conserved cysteine residue and two from a pyranopterin dithiolene cofactor), one axial oxo group and an equatorial OH or OH2 group whose identity varies during the catalytic cycle.
Catalysis in SOEs is a complex multistep process, which involves the synchronization of various chemical events, such as electron and proton transfer and the formation of substrate and product intermediates whose properties are finely tuned by the protein environment. Characterizing them is challenging due to enzyme turnover. For instance, the formation of a covalently bound Mo(IV)–O–SO3 reaction intermediate has been proposed following substrate binding (Fig. S1, ESI†),4,8–10 which is supported by recent theoretical investigations.11 However, no such intermediate could be trapped so far. Three high-resolution structures of SOEs have been solved with either a SO42− (in chicken SO (cSO)12 and in Starkeya novella SDH (SorAB)),13 or a SO32− (in the C185S variant of cSO)14 molecule close to their active site. However, in all of them, the SO42−/SO32− oxygen atom closest to the Mo is found at distances ≥3.0 Å that preclude direct coordination to the Mo atom. On the other hand, evidences for direct coordination of SO32− rather than SO42− to the Mo atom have been provided in plant15 and human SOs,16–18 as well as in a variant of SorAB (R55Q)19 through detailed EPR characterizations of non-catalytic sulfite Mo(V) species (renamed “blocked”).7,20
In this context, structural and functional studies using substrate or product analogues are crucial for providing detailed insights into SOEs’ functioning. So far, direct binding of the product analogues PO43− and AsO43− to the Mo(V) ion has been evidenced in vertebrate SOs using EPR and EXAFS spectroscopies,4,21–23 while structural evidence is still lacking.
In this work, we show that SDH from Thermus thermophilus (TtSDH) can serve as a model enzyme to remedy this lack of detailed structural information on the binding of the product analogue PO43− at the Mo active site in SOEs. TtSDH is a homodimeric enzyme containing only the Mo redox center.24 It interacts with a diheme cytochrome c550, which acts as physiological electron acceptor. The electrons are then shuttled to O2 reductases through cytochrome c552, linking SO32− oxidation to the aerobic respiration pathway.24,25 Here, TtSDH was crystallized in the presence of 1.8 M Na3PO4 and its structure solved by X-ray crystallography (Table S1, ESI†). Like other SOEs, the overall structure of TtSDH is organized in two domains: a catalytic domain containing the Moco active site and a dimerization domain (Fig. S2, ESI†). The structure features a well-resolved electron density in the active site of the protein consistent with the presence of the Moco. Interestingly, where the available structures of SOEs normally show an oxo-ligand coordinated to the Mo atom at the equatorial position, an extended positive tetrahedral shaped electron density was observed adjacent to the Mo atom in TtSDH (Fig. 1). This could arise either from SO42− present in the culture media or, more likely, from the phosphate present in the crystallization solution at high concentration. As both molecules are indistinguishable based solely on the electron density map, we used EPR techniques to further assess the nature of the anion coordinated to the Mo atom.
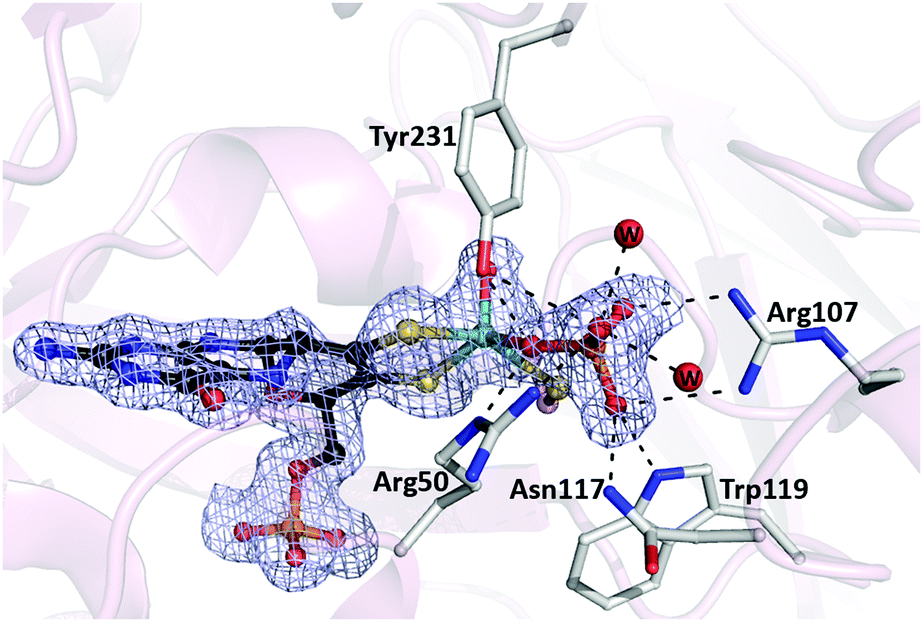 | ||
| Fig. 1 Active site of TtSDH. Fo–Fc omit map contoured at 3σ (light blue mesh) showing the Moco with the phosphate molecule directly coordinated to the Mo atom. Residues involved in phosphate binding are shown in sticks. Water molecules involved in H-bonds with the phosphate molecule are labeled w. All H-bonds are shown as dashed lines. | ||
The X-band (∼9 GHz) CW EPR spectrum of a redox-poised sample of TtSDH containing 1.8 M Na3PO4 at pH 5.7 features an intense anisotropic signature with resolved splittings that lead to overlapping EPR lines in the high field region of the spectrum (Fig. 2). X- and Q-band (∼34 GHz) (Fig. S3, ESI†) EPR spectra are well simulated using a similar set of parameters corresponding to a single rhombic species with g-values (g1, g2, g3) = (1.9966, 1.9700, 1.9622) and experiencing a strong magnetic interaction with a nucleus with spin value I = ½. This signal is assigned to a 4d1 Mo(V) atom in a low symmetry coordination sphere on the basis of its g-values, its relaxation properties and the detected lower intensity hyperfine lines from the I = 5/2 95,97Mo isotopes (25% natural abundance) that superimpose on the main spectral contribution.26 Simulations further show that the resolved splittings are well reproduced using a nearly axial hyperfine tensor with principal values (A1, A2, A3) = (22, 21, 29) MHz and assuming collinear g- and hf-main axes (Fig. 2). The identity of the I = ½ coupled nucleus was determined using HYSCORE spectroscopy.
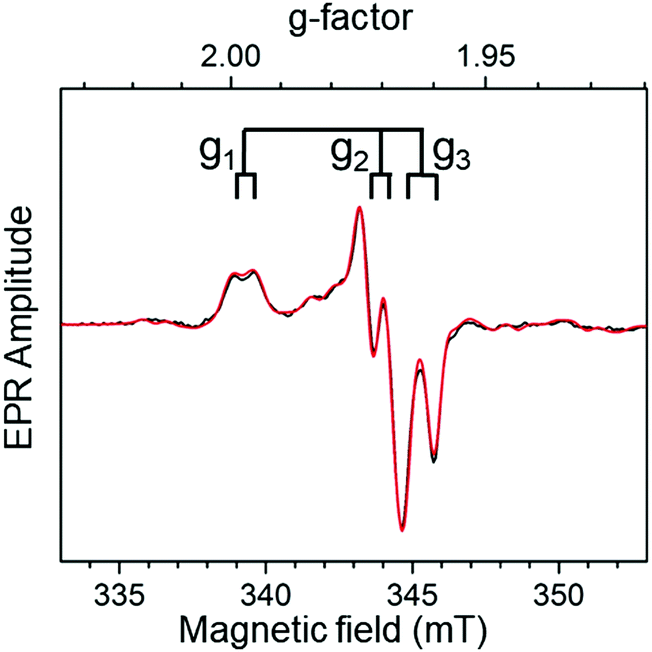 | ||
| Fig. 2 X-Band (∼9.4803 GHz) EPR spectrum of the Mo(V) species in TtSDH incubated with 1.8 M Na3PO4. The experimental spectrum (black trace) is shown together with its simulation (red trace) performed using the parameters provided in the ESI† text and Table S3. Other experimental conditions are also given in the ESI.† | ||
The (−, +) quadrant of the Mo(V) HYSCORE spectrum measured at the g2 field position is shown in Fig. 3. It features a pair of intense cross-peaks produced by a strongly coupled nucleus with hyperfine coupling constant A satisfying the relationship A > 2νI (νI is the nuclear Zeeman frequency). The correlated nuclear frequencies with maxima around ν± ≈ (18.5, 6.6) MHz are separated by twice the 31P Zeeman frequency (νP ≈ 6.0 MHz), and are thus assigned to a 31P nucleus. The shape and the position of these cross-peaks are well simulated assuming an axially symmetric 31P hyperfine tensor with isotropic and anisotropic components (|Aiso|, |T|) = (26, 3.6) MHz, respectively. These values are consistent with those inferred from the simulation of the cw EPR spectra (i.e. (|Aiso|, |T|) ≈ (24, 2.5) MHz). This unambiguously shows that the HYSCORE cross-peaks analyzed above and the splitting resolved on the X-band cw EPR spectrum originate from a single strongly coupled 31P nucleus, assigned to a Mo–phosphate adduct. Indeed, the magnitude of the 31P Aiso value is larger7,27–29 or comparable23 to those estimated from previous pulsed EPR experiments in vertebrate SOs and assigned to phosphate coordinated species. Distributed 31P Aiso values ranging from ∼0 to ∼20 MHz have been estimated in cSO.29 The absence of such a wide distribution in TtSDH, comparatively suggests a more rigid environment around the bound phosphate molecule. Eventually, similar spectra were recorded using a TtSDH sample incubated with a lower (i.e. 70 mM) Na3PO4 concentration as typically used in previous SOs studies (Fig. S4, ESI†).21,22,27
 | ||
| Fig. 3 X-Band (∼9.6980 GHz) HYSCORE spectrum of the Mo(V) species in TtSDH incubated with 1.8 M Na3PO4. A simulation depicted as red contour plot is superimposed to the (−, +) quadrant of the frequency-domain HYSCORE spectrum. 31P and 13C hyperfine interaction tensors used for spectral simulation are given in the text and in the ESI,† respectively. Double arrows schematically indicate double the 31P and 13C Larmor frequencies (referred to as 2νP and 2νC, respectively). Experimental parameters are given in the ESI,† magnetic field value, 351.7 mT. | ||
Upon placement of phosphate in the electron density map and subsequent structure refinement, the product analogue clearly appears to be directly coordinated to the Mo atom, with one of its oxygens being at the position where the equatorial oxo-ligand is normally observed (Fig. 1). This phosphate oxygen is only at 2.16 Å distance from the Mo atom, which is within the known range for Mo–phosphate complexes.30
Such a direct coordination is further supported by DFT calculations of EPR parameters performed on large (∼670 atoms) molecular models of the TtSDH active site based on atomic coordinates from the phosphate-bound structure (Fig. S5, ESI†). The three geometry-optimized models obtained upon consideration of the different possible protonation states of the coordinated phosphate molecule are referred to as models 1a-[HPO4]2− and 1b-[HPO4]2− (with monoprotonated phosphate) and model 2-[H2PO4]− (with diprotonated phosphate). Models 1a and 1b correspond to two energy minima of the bound HPO42− placing the phosphate proton with two different orientations (Fig. S6A and B, ESI†). Overall, the three optimized structures are very similar to each other and only subtle structural changes are resolved compared to the original X-ray structure (Table S2, ESI†). Regarding the Mo(V) g-tensor, the phosphate protonation state mainly affects the g1 value which increases significantly when the substrate analogue is deprotonated (Table S3, ESI†). Best agreement between the calculated and experimentally determined g-values is achieved for model 1a, suggesting that the phosphate molecule is in the monoprotonated form in the conditions used for EPR analyses (Table S3, ESI†). In addition, 31P Aiso values in the 15–40 MHz range are calculated for the three models, in agreement with the value of 24 MHz measured herein. It is noteworthy that the experimental and the calculated Oax–Mo–O–P dihedral angle values (defined according to counter clockwise rotation of the Mo–O–P fragment about the Mo–O bond) range from −59° to −70° (Table S2, ESI†). These values place the phosphorus atom close to the equatorial plane where the unpaired electron primarily resides, thereby contributing to the relatively large 31P Aiso value measured in this work.4,7 Overall, correlation of the experimental EPR results with extended DFT calculations on structural models that include surroundings and interactions of the active site provides further support for direct phosphate coordination to the molybdenum active site in TtSDH.
The identification of a phosphate-bound Mo(V) species in a SDH is unprecedented. A proposed prerequisite for the formation of phosphate-inhibited species in SOs is the ability of the Mo center to adopt a so-called “low pH ligand environment” when these enzymes are prepared at low pH (≤7.5) and high chloride concentration (∼100 mM).31 This environment involves the possibility of the equatorial hydroxyl group to adopt a specific orientation that differs to that of the “high pH-type” occurring in all known SOEs.4 Notably, between the two possible orientations proposed from EPR and DFT analyses, the one defined by an Oax–Mo–O–H dihedral angle of −60° for the low pH Mo(V) form in SOs is similar to the corresponding one of the phosphate moiety evidenced in the present work. Interestingly, low pH Mo(V) species have also never been reported in SDHs.6 This apparent peculiarity of prokaryotic SOEs could be due to the low number of SDHs investigated in details so far using EPR methods. The possibility to generate such a low pH Mo(V) in TtSDH will therefore be addressed in future investigations.
The herein characterized HPO42−-bound complex is likely to mimic the covalently bound Mo(IV)–O–SO3 reaction intermediate proposed to occur in the catalytic cycle of SOEs after substrate binding and oxidation,4,8,9 providing new insights into substrate/product recognition in their active site. Indeed, HPO42− displays a similar volume and the same charge as SO42−. Moreover, our modelling studies reveal that the HPO42− proton does not form any additional H-bond interaction (Fig. S5, ESI†). On the other hand, our high-resolution structure shows that the phosphate molecule is tightly bound by a dense H-bond network originating from Arg50, Arg107, Asn117, Trp119 and Tyr231 side chains, creating a favorable electrostatic environment for anion binding (Fig. 1). Two water molecules participate in this network by bridging H-bonds to Asn117 main chain and Arg230 and Tyr367 side chains. Besides their involvement in stabilizing the reaction intermediate, these H-bond interactions are predicted to contribute to lower activation barriers.11,32
Comparison of the TtSDH structure with those of cSO and SorAB featuring SO42− molecules close to the Mo atom reveals significant differences. Indeed, in cSO, the sulfate molecule in the active site is located at 4.6 Å (nearest SO42− oxygen) to the Mo atom, which itself still remains coordinated to the equatorial oxygen ligand.12 Replacement by Gln of Arg138, proposed as crucial for catalysis, does not affect the binding mode but increases the distance between the SO42− molecule and the Mo atom to 5.5 Å.12 By contrast, a structure of SorAB with SO42− bound at the active site could only be obtained when the equivalent Arg residue (Arg55) was replaced by Met despite high SO42− concentration in the crystallization solution (i.e. 2.2 M).13 In this R55M variant, the Met residue side chain has been shown to adopt a different orientation compared to the Arg residue, leaving space for the SO42− molecule to approach the Mo atom at a distance of 3.0 Å, however, without being coordinated. While no conformational change of active site residues is observed in the R55M variant of SorAB upon SO42− binding, an Arg residue (Arg450) which is solvent exposed in the SO42− free structure of the cSO has been shown to point towards the active site in the presence of SO42−. This Arg residue participates in the H-bond network of what seems to be a leaving SO42− molecule and, thus, has been proposed to play a gating role in cSO.13 In contrast, the side chain of Arg 365 in TtSDH, which occupies the same position as Arg 450 in cSO, is solvent exposed in our phosphate-bound structure, excluding a similar role in TtSDH for this residue. Rather, a further Arg residue in TtSDH (Arg230) is found to be oriented towards the HPO42− molecule at 4.1 Å, suggesting a role in directing the substrate/product to or from the active site. The precise role of this arginine therefore deserves to be assessed using site-directed mutagenesis.
In conclusion, by combining X-ray crystallography, CW and pulsed EPR spectroscopy and DFT modelling, we provide clear evidence for direct coordination of HPO42− to the Mo atom at the active site of a SOE, revealing the molecular bases for the stabilization of the covalently bound reaction intermediate through an intricate H-bond network. The present work opens up new perspectives for future detailed investigations on the biological conversion mechanism of SO32− into SO42− in molybdoenzymes.
We acknowledge Diamond Light Source for time on Beamline I24 under Proposal [mx17293]. This work was supported by Aix Marseille Univ and CNRS. Support from the Libyan ministry of higher education and scientific research for S. A. doctoral program is acknowledged. The authors are also grateful to the EPR facilities available at the French EPR network (RENARD, IR CNRS 3443) and the Aix-Marseille University EPR center. DFT calculations were performed by using computing resources from the “Centre Régional de Compétences en Modélisation Moléculaire” (CRCMM, Marseille).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- R. M. Garrett, J. L. Johnson, T. N. Graf, A. Feigenbaum and K. V. Rajagopalan, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6394–6398 CrossRef CAS PubMed.
- J. Simon and P. M. Kroneck, Adv. Microb. Physiol., 2013, 62, 45–117 CrossRef CAS PubMed.
- J. H. Enemark, Dalton Trans., 2017, 46, 13202–13210 RSC.
- M. J. Pushie and G. N. George, Coord. Chem. Rev., 2011, 255, 1055–1084 CrossRef CAS.
- U. Kappler and J. H. Enemark, J. Biol. Inorg. Chem., 2015, 20, 253–264 CrossRef CAS PubMed.
- E. L. Klein, A. V. Astashkin, A. M. Raitsimring and J. H. Enemark, Coord. Chem. Rev., 2013, 257, 110–118 CrossRef CAS PubMed.
- R. R. Crichton, in Biological Inorganic Chemistry, ed. R. R. Crichton, Elsevier, Amsterdam, 2008, pp. 279–296 DOI:10.1016/B978-044452740-0.50017-X.
- J. H. Enemark, A. V. Astashkin and A. M. Raitsimring, Biochem. Soc. Trans., 2008, 36, 1129–1133 CrossRef CAS PubMed.
- R. Hille, J. Biol. Inorg. Chem., 1997, 2, 804–809 CrossRef CAS.
- O. Caldararu, M. Feldt, D. Cioloboc, M. C. van Severen, K. Starke, R. A. Mata, E. Nordlander and U. Ryde, Sci. Rep., 2018, 8, 4684 CrossRef PubMed.
- E. Karakas, H. L. Wilson, T. N. Graf, S. Xiang, S. Jaramillo-Buswuets, K. V. Rajagopalan and C. Kisker, J. Biol. Chem., 2005, 280, 33506–33515 CrossRef CAS PubMed.
- S. Bailey, T. Rapson, K. Johnson-Winters, A. V. Astashkin, J. H. Enemark and U. Kappler, J. Biol. Chem., 2009, 284, 2053–2063 CrossRef CAS PubMed.
- J. A. Qiu, H. L. Wilson, M. J. Pushie, C. Kisker, G. N. George and K. V. Rajagopalan, Biochemistry, 2010, 49, 3989–4000 CrossRef CAS PubMed.
- A. V. Astashkin, K. Johnson-Winters, E. L. Klein, R. S. Byrne, R. Hille, A. M. Raitsimring and J. H. Enemark, J. Am. Chem. Soc., 2007, 129, 14800–14810 CrossRef CAS PubMed.
- A. V. Astashkin, K. Johnson-Winters, E. L. Klein, C. Feng, H. L. Wilson, K. V. Rajagopalan, A. M. Raitsimring and J. H. Enemark, J. Am. Chem. Soc., 2008, 130, 8471–8480 CrossRef CAS PubMed.
- A. Rajapakshe, K. Johnson-Winters, A. R. Nordstrom, K. T. Meyers, S. Emesh, A. V. Astashkin and J. H. Enemark, Biochemistry, 2010, 49, 5154–5159 CrossRef CAS PubMed.
- E. L. Klein, A. A. Belaidi, A. M. Raitsimring, A. C. Davis, T. Kramer, A. V. Astashkin, F. Neese, G. Schwarz and J. H. Enemark, Inorg. Chem., 2014, 53, 961–971 CrossRef CAS PubMed.
- T. D. Rapson, A. V. Astashkin, K. Johnson-Winters, P. V. Bernhardt, U. Kappler, A. M. Raitsimring and J. H. Enemark, J. Biol. Inorg. Chem., 2010, 15, 505–514 CrossRef CAS PubMed.
- R. C. Bray, M. T. Lamy, S. Gutteridge and T. Wilkinson, Biochem. J., 1982, 201, 241–243 CrossRef CAS PubMed.
- G. N. George, R. M. Garrett, T. Graf, R. C. Prince and K. V. Rajagopalan, J. Am. Chem. Soc., 1998, 120, 4522–4523 CrossRef CAS.
- S. Gutteridge, M. T. Lamy and R. C. Bray, Biochem. J., 1980, 191, 285–288 CrossRef CAS PubMed.
- E. L. Klein, A. M. Raitsimring, A. V. Astashkin, A. Rajapakshe, K. Johnson-Winters, A. R. Arnold, A. Potapov, D. Goldfarb and J. H. Enemark, Inorg. Chem., 2012, 51, 1408–1418 CrossRef CAS PubMed.
- S. Robin, M. Arese, E. Forte, P. Sarti, A. Giuffre and T. Soulimane, J. Bacteriol., 2011, 193, 3988–3997 CrossRef CAS PubMed.
- S. Robin, M. Arese, E. Forte, P. Sarti, O. Kolaj-Robin, A. Giuffre and T. Soulimane, PLoS One, 2013, 8, e55129 CrossRef CAS PubMed.
- S. Grimaldi, F. Biaso, B. Burlat and B. Guigliarelli, in Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations, ed. R. Hille, C. Schulzke and M. L. Kirk, The Royal Society of Chemistry, 2016, pp. 68–120 Search PubMed.
- M. T. Lamy, S. Gutteridge and R. C. Bray, Biochem. J., 1980, 185, 397–403 CrossRef CAS PubMed.
- A. M. Raitsimring, A. Pacheco and J. H. Enemark, J. Am. Chem. Soc., 1998, 120, 11263–11278 CrossRef CAS.
- A. Pacheco, P. Basu, P. Borbat, A. M. Raitsimring and J. H. Enemark, Inorg. Chem., 1996, 35, 7001–7008 CrossRef CAS PubMed.
- J. Fischer, L. Ricard and P. Toledano, J. Chem. Soc., Dalton Trans., 1974, 941–946, 10.1039/dt9740000941.
- U. Kappler, B. Bennett, J. Rethmeier, G. Schwarz, R. Deutzmann, A. G. McEwan and C. Dahl, J. Biol. Chem., 2000, 275, 13202–13212 CrossRef CAS PubMed.
- E. Hernandez-Marin and T. Ziegler, Inorg. Chem., 2009, 48, 1323–1333 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc03634j |
| ‡ Present address: Azzaytuna University, Faculty of Science, Biology Department, Tarhuna City, Libya. |
| This journal is © The Royal Society of Chemistry 2020 |