
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO–I–O halogen bond of halonium ions†
Sofia
Lindblad
 a,
Flóra Boróka
Németh
b,
Tamás
Földes
b,
Alan
Vanderkooy
a,
Imre
Pápai
b and
Máté
Erdélyi
*a
a,
Flóra Boróka
Németh
b,
Tamás
Földes
b,
Alan
Vanderkooy
a,
Imre
Pápai
b and
Máté
Erdélyi
*a
aDepartment of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. E-mail: mate.erdelyi@kemi.uu.se
bInstitute of Organic Chemistry, Research Center for Natural Sciences, Budapest, Hungary
First published on 13th July 2020
Abstract
The reactivity of halonium ions is conveniently modulated by three-center, four-electron halogen bonds. Such stabilized halonium complexes are valuable reagents for oxidations and halofunctionalization reactions. We report the first example of the stabilization of a halenium ion in a three-center, four-electron halogen bond with two oxygen ligands. The influence of electron density and solvent on the stability of the complexes is assessed. O–I–O halogen bond complexes are applicable as synthetic reagents and as supramolecular synthons.
A halogen bond is the attractive interaction of an electrophilic area of a halogen with a nucleophile.1 It was first observed in 1863 by Guthrie,2 but only began to receive significant attention towards the end of the 20th century. Today, halogen bonding is one of the fastest growing research topics in chemistry3 with applications for example in crystal engineering,4,5 drug design,6–9 material sciences,10–12 and organic synthesis.13–15
A halenium ion16 (X+) has an electron depleted p-orbital that can interact simultaneously with two Lewis bases, forming a linear three-center, four-electron (3c4e) halogen bond.17 Due to their electron deficiency, halonium ions are strong halogen bond donors. The energy range of halogen bonds overlaps with that of hydrogen bonds,18 with the 3c4e halogen bonds being among the strongest secondary interactions (up to 120 kJ mol−1).17 In addition to the 3c4e bonds19 in trihalide anions,20 3c4e halogen bond complexes have previously been formed using nitrogen,21–23 sulphur,24–26 selenium,27,28 tellurium,29 and mixed nitrogen and oxygen electron donors30–32 (Fig. S1, ESI†). Similarly to analogous short, strong hydrogen bonds (SSHB), which are of interest for stabilizing reaction intermediates and transition states,33,34 3c4e halogen bond complexes are applicable as synthetic reagents and in the design of complex supramolecular structures.17,26,35–39
We report herein the first [O–I–O]+-type 3c4e halogen bond complexes, employing two oxygen donor ligands for the stabilization of a reactive halenium ion. This is significant as investigations of halogen bonding applying oxygen ligands as Lewis bases have been scarce,40 and 3c4e halogen bonding with two oxygen ligands has so far been scarcely explored.17,51 Our study provides new insights into both halogen bonding and 3c4e bonds. Additionally, [O–I–O]+ complexes are of interest for the development of new supramolecular synthons and of improved reagents for halofunctionalization and oxidation reactions.
Due to the formal negative charge on the oxygen, we anticipated pyridyl N-oxides to be suitable Lewis bases for the formation of an [O–I–O]+ 3c4e halogen bond. The complex (1-OMe)2-I (Fig. 1 and Table 1) was synthesized from the electron rich 4-methoxypyridine N-oxide (1-OMe) following a protocol previously applied for the generation of the analogous [N–I–N]+ halogen bond complexes23 (ESI†). Formation of the 3c4e halogen bond was confirmed by the −31.2 ppm 15N NMR coordination shift in dry CD3CN, as detected on the nitrogen of the 1-OMe ligand (δ15Ncoord = δ15Ncomplex − δ15Nligand, Table 1).
 | ||
| Fig. 1 The 15N NMR chemical shifts of 4-methoxypyridine N-oxide (1-OMe, pink) and of its silver(I) (black, (1-OMe)2-Ag), iodine (green, (1-OMe)2-I2), iodine(I) (red, (1-OMe)2-I) and proton (blue, (1-OMe)-H) complexes were acquired by 1H,15N HMBC NMR (CD3CN, 500/51 MHz), with detection on the proton meta to the pyridine nitrogen (H-3) of the complexes. | ||
| Structure | δ 15Na (ppm) | δ 15Ncalcd (ppm) | ΔGrele (kcal mol−1) | r(O–X)f [Å] | σ(N–O–X)g [°] | |
|---|---|---|---|---|---|---|
| a Measured by 1H,15N HMBC. b Measured in CD3CN. c Measured in CD2Cl2. d Computed using DFT with MeCN as solvent. e Relative stabilities of (OR)2–X (X = I, Ag) and (OR)⋯I2 complexes are defined as Gibbs free energies of reactions [Py–X–Py]+ + 2OR → [RO–X–OR]+ + 2Py and OR + I2 → RO⋯I2, respectively. f Computed O–Ag and O–I bond distances. g Computed N–O–Ag and N–O–I bond angles, see the ESI for computational details. | ||||||
| (1-OMe)2-I |

|
−134.2b | −143.5 | −2.7 | 2.22 | 114.0 |
| (1-OMe)⋯I2 |

|
−122.8b | −136.5 | −2.0 | 2.36 | 112.9 |
| (1-OMe)2-Ag |

|
−105.1b | −131.4 | 4.5 | 2.12 | 119.3 |
| 1-OMe |

|
−103.0b | −98.8 | |||
| (1-Me)2-I |

|
−121.2,b −119.4c | −129.0 | 1.9 | 2.22 | 113.5 |
| (1-Me)⋯I2 |

|
−102.2,b −98.9c | −120.1 | −0.4 | 2.39 | 112.7 |
| (1-Me)2-Ag |
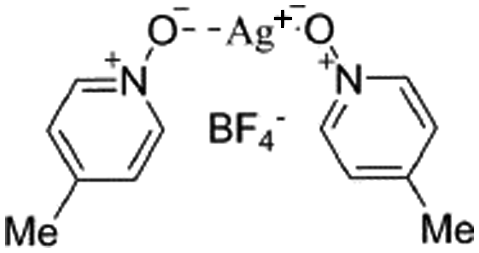
|
−92.0b | −116.6 | 7.8 | 2.12 | 124.5 |
| 1-Me |

|
−91.7,b −92.1c | −86.6 | |||
This δ15Ncoord is in reasonable agreement with that predicted by DFT, and is significantly smaller than that of the analogous [N–I–N]+ complex of iodine(I) formed with pyridine ((Py)2-I, δ15Ncoord = −109.0 ppm).41 Computations predict the (1-OMe)2-I complex to be slightly more stable than (Py)2-I (ΔGrel = −2.7 kcal mol−1, Table 1). The smaller |δ15Ncoord| yet larger stability of (1-OMe)2-I as compared to (Py)2-I is not a contradiction because the iodine(I) of the former complex does not coordinate directly to the nitrogen but to the N-oxide oxygen. The larger |δ15Ncoord| of (1-OMe)2-I as compared to (1-OMe)2-Ag (δ15Ncoord = −2.1 ppm, Fig. 1 and Table 1) is explained by CD3CN coordination to silver(I) but not to the iodine(I) of the 3c4e complex (Fig. S77, ESI†).42,43 A previous study of silver(I) pyridine-N-oxide complexes reported the formation of various coordination complexes, such as [(Py-O)2-Ag](NO3), [(Py-O)2-Ag](OMs), [(Py-O)3-Ag](OTf2N), [(Py-O)3-Ag](OTf), [(Py-O)8-Ag3](OTf)3 in the solid state.44 This further confirms that silver(I) may form multicoordinate species. (1-OMe)2-Ag is likely in a ligand exchange equilibrium with CD3CN ΔG < 2 kcal mol−1, (Table S3, ESI†), which explains the deviation of the small |δ15Ncoord| of (1-OMe)2-Ag from the value predicted by DFT (Table 1 and Fig. S77, ESI†). Hence, in contrast to [N–Ag–N]+ complexes,42 the [O–Ag–O]+ complex (1-OMe)2-Ag is to a large extent dissociated in CD3CN. Upon mixing 1-OMe with iodine in CD3CN, at the same concentrations as used to form (1-OMe)2-I, the coordination shift was consistent with the formation of a weak, conventional 1-OMe⋯I2 halogen bond (δ15Ncoord = −11.4 ppm), which is also supported by the computed exergonicity of association (ΔGrel = −2.0 kcal mol−1). Although the coordination shift is not necessarily a quantitative indicator for bond strength,41 the 63% smaller |δ15Ncoord| of 1-OMe⋯I2 as compared to (1-OMe)2-I is a qualitative reflection of the relative strengths of the conventional and the 3c4e halogen bonds with the same Lewis base, 1-OMe.
The complex (1-OMe)2-I was applicable for iodocyclization of 4-penten-1-ol to form 2-(iodomethyl)tetrahydrofuran, with a comparable reaction rate to Barluenga's reagent, [bis(pyridine)-iodine(I)]+ BF4−, indicating the potential applicability of [O–I–O]+ 3c4e halogen bond complexes as synthetic reagents (Fig. 2).38 This suggestion is corroborated by the higher stability of (1-OMe)2-I as compared to Barluenga's reagent13,14 (ΔGrel, Table 1), as calculated by DFT. A higher stability may result in improved reaction control and simpler reagent storage, for example.
 | ||
| Fig. 2 Complex (1-OMe)2-I was applicable as a halenium transfer reagent for the iodocyclization of 4-penten-1-ol (see the ESI† for details). | ||
The somewhat less electron rich 1-Me ligand also provided a 3c4e halogen bond complex, (1-Me)2-I in dry CD2Cl2, with its formation being indicated by the observation of δ15Ncoord = −27.7 ppm (Table 1). This complex was applicable as halenium transfer reagent in the halocyclization of 4-penten-1-ol, similar to (1-OMe)2-I (Fig. 2). When forming (1-Me)2-I in CD3CN, we observed some hydrolysis yielding 1-Me-H, i.e. protonated N-oxide (Fig. S47–S52, ESI†). This is explained by the higher residual water content of acetonitrile as compared to dichloromethane,45 unlikely to be a consequence of different solvent polarities.46 The (1-Me)2-I and (1-OMe)2-I complexes were stable at room temperature in solution under dry conditions, yet showed a larger moisture sensitivity than the corresponding [bis(pyridine)iodine(I)]+-type N–I–N halogen bond complexes.46,47
Attempts to form an [O–I–O]+ halogen bond complex with dibenzofuran, a weaker Lewis base, did not succeed (Fig. S63–S67, ESI†). This was reflected by the lack of silver iodide precipitation upon addition of AgBF4 and I2 to the acetonitrile solution of dibenzofuran, the lack of 1H NMR chemical shift changes throughout the experiment, and the DFT predicted instability of the corresponding iodine(I) complex (see the ESI† for details). This observation accentuates the importance of the identity of the Lewis base in the formation of stable halogen bond complexes.
The DFT optimized geometries of (1-OMe)2-I+ and (1-Me)2-I+ cations, shown in Fig. 3, possess the expected linear [O–I–O]+ halogen bonds. It should be emphasized that the overall geometry vastly differs from the linear [N–I–N]+ complexes formed by 4-methoxy- and 4-methylpyridine.41 The overall bent geometries present opportunities for constructing alternate supramolecular architectures when using [O–X–O]+ 3c4e complexes of pyridine N-oxides, as compared to the [N–X–N]+ complexes formed by the corresponding pyridines.
 | ||
| Fig. 3 Computationally optimized structures of the iodonium complexes with 1-OMe and 1-Me. Structures were optimized by DFT at ωB97X-D/Def2SVP level. For details on the negligible influence of the BF4− counterion, see the ESI† and ref. 43. Selected bond distances are in Å, bond angles in degrees (φ refers to dihedral angle N–O⋯O–N). | ||
Rotation about the O–I–O halogen bonds is practically free, as indicated by the marginal energy barrier, less than 0.3 kcal mol−1, predicted with DFT. The calculated O–I bonds show 37.2% shortening of the distance of the involved atoms as compared to the sum of their van der Waals radii (RXB = 0.628, Bondi's radii,48,49Table 1).17 This is comparable to the previously reported RXB = 0.612–0.654 of the analogous [N–I–N]+ complexes,22,41,45,50 and the RXB = 0.662 of the [N–I–O]− halogen bond complexes (I–O distance 2.316 Å).30 This further supports the formation of true 3c4e bonds17,22 for (1-OMe)2-I and (1-Me)2-I.
In conclusion, we report the first examples of 3c4e [O–X–O]+ halogen bond complexes. N-Oxides are shown to be privileged oxygen-donor ligands, as no such complex could be observed using an ether as Lewis base. Our data demonstrates the immense impact of the electron density of the electron donor on halogen bonding, with a 4.6 kcal mol−1 relative stability decrease predicted upon a methoxy to methyl substitution, which was confirmed experimentally by the lower stability of (1-Me)2-I, in contrast to (1-OMe)2-I, in CD3CN. The higher stability of (1-Me)2-I in CD2Cl2 as compared to CD3CN further reveals the importance of the solvent in the preparation of stabilized halonium complexes. This is of significance as Barluenga's reagent, for instance, was shown to be more reactive in CD3CN as compared to CD2Cl2.14 The overall bent geometry of these complexes makes the [O–I–O]+ bond a potential synthon that will result in different supramolecular architectures than those formed by the [N–I–N]+ complexes of analogous pyridines. Furthermore, the reported [O–I–O]+ halogen bond complexes are proven to be applicable as synthetic reagents, and are expected to be of interest in the development of future oxidation and halofunctionalization reagents. Introducing the [O–X–O]+ motif, this work expands the scope of ligands known to be competent in the formation of 3c4e halogen bond complexes. Their further studies will provide novel fundamental insights into both the halogen bond and the 3c4e bonding phenomena.
This project made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy. We thank the Swedish Research Council (2016-03602) and FORMAS (2017-01173) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CAS.
- F. Guthrie, J. Am. Chem. Soc., 1863, 16, 239 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed.
- J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225 CrossRef CAS PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061 CrossRef CAS PubMed.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854 CrossRef CAS PubMed.
- Y. Lu, Y. Wang and W. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543 RSC.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363 CrossRef CAS PubMed.
- P. Metrangolo, G. Resnati, T. Pilati, R. Liantonio and F. Meyer, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1 CrossRef CAS.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed.
- J. Barluenga, J. M. González, P. J. Campos and G. Asensio, Angew. Chem. Int. Ed. Engl., 1985, 24, 319 CrossRef.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Chem. – Eur. J., 2004, 10, 4206 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622 CrossRef CAS.
- Note on nomenclature: X+ stands for hypovalent (6e) halenium, and [R–X–R]+ for hypervalent (10e) halonium ion; X+ is a halogen bond donor with a Lewis basic empty p orbital and [R–X–R]+ a halogen bonded complex.
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688 RSC.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547 RSC.
- A. C. Reiersolmoen, S. Battaglia, S. Oien-Odegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdelyi, Chem. Sci., 2020 10.1039/D0SC02076A.
- P. Deplano, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, Coord. Chem. Rev., 1999, 188, 71 CrossRef CAS.
- A.-C. C. Carlsson, A. X. Veiga and M. Erdélyi, Top. Curr. Chem., 2015, 359, 49 CrossRef CAS PubMed.
- S. B. Hakkert and M. Erdelyi, J. Phys. Org. Chem., 2015, 28, 226 CrossRef CAS.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706 CrossRef CAS PubMed.
- L. Koskinen, S. Jääskeläinen, P. Hirva and M. Haukka, Cryst. Growth Des., 2015, 15, 1160 CrossRef CAS.
- L. Koskinen, P. Hirva, A. Hasu, S. Jääskeläinen, J. Koivistoinen, M. Pettersson and M. Haukka, CrystEngComm, 2015, 17, 2718 RSC.
- L. Koskinen, P. Hirva, E. Kalenius, S. Jääskeläinen, K. Rissanen and M. Haukka, CrystEngComm, 2015, 17, 1231 RSC.
- E. Seppälä, F. Ruthe, J. Jeske, W.-W. du Mont and P. G. Jones, Chem. Commun., 1999, 1471 RSC.
- W.-W. du Mont, M. Bätcher, C. Daniliuc, F. A. Devillanova, C. Druckenbrodt, J. Jeske, P. G. Jones, V. Lippolis, F. Ruthe and E. Seppälä, Eur. J. Inorg. Chem., 2008, 4562 CrossRef CAS.
- G. M. de Oliveira, E. Faoro and E. S. Lang, Inorg. Chem., 2009, 48, 4607 CrossRef.
- R. Puttreddy, O. Jurček, S. Bhowmik, T. Mäkelä and K. Rissanen, Chem. Commun., 2016, 52, 2338 RSC.
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigue, Faraday Discuss., 2017, 203, 389 RSC.
- R. Puttreddy, J. M. Rautiainen, T. Makela and K. Rissanen, Angew. Chem. Int. Ed., 2019, 58, 18610 CrossRef CAS PubMed.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91 RSC.
- J. A. Gerlt, M. M. Kreevoy, W. W. Cleland and P. A. Frey, Chem. Biol., 1997, 4, 259 CrossRef CAS PubMed.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343 RSC.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714 CrossRef CAS.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem. Int. Ed., 2016, 55, 14033 CrossRef CAS.
- A. Vanderkooy, A. K. Gupta, T. Foldes, S. Lindblad, A. Orthaber, I. Papai and M. Erdelyi, Angew. Chem. Int. Ed., 2019, 58, 9012 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861 CAS.
- V. Nemec, L. Fotovic, T. Vitasovic and D. Cincic, CrystEngComm, 2019, 21, 3251 RSC.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853 CrossRef CAS PubMed.
- R. Kleinmaier, S. Arenz, A. Karim, A.-C. C. Carlsson and M. Erdélyi, Magn. Reson. Chem., 2013, 51, 46 CrossRef CAS.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topic, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdelyi, Chem. Sci., 2015, 6, 3746 RSC.
- J. Sniekers, N. R. Brooks, S. Schaltin, L. Van Meervelt, J. Fransaer and K. Binnemans, Dalton Trans., 2014, 43, 1589 RSC.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351 CrossRef CAS PubMed.
- A.-C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Gräfenstein and M. Erdelyi, CrystEngComm, 2013, 15, 3087 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdelyi, Chem. Commun., 2012, 48, 1458 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- O. Hassel and H. Hope, Acta Chem. Scand., 1961, 15, 407 CrossRef CAS.
- M. Kandrnálová, Z. Kokan, V. Havel, M. Nečas and V. Šindelář, Angew. Chem. Int. Ed., 2019, 58, 18182 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and details on synthesis and computation. See DOI: 10.1039/d0cc03513k |
| This journal is © The Royal Society of Chemistry 2020 |