
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroswitching of the single-molecule magnet behaviour in an octahedral spin crossover cobalt(II) complex with a redox-active pyridinediimine ligand†‡
Renato
Rabelo
a,
Luminita
Toma
*a,
Nicolás
Moliner
 a,
Miguel
Julve
a,
Francesc
Lloret
a,
Jorge
Pasán
b,
Catalina
Ruiz-Pérez
b,
Rafael
Ruiz-García
a and
Joan
Cano
*a
a,
Miguel
Julve
a,
Francesc
Lloret
a,
Jorge
Pasán
b,
Catalina
Ruiz-Pérez
b,
Rafael
Ruiz-García
a and
Joan
Cano
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, 46980 Paterna, València, Spain. E-mail: luminita.toma@uv.es; joan.cano@uv.es
bLaboratorio de Rayos X y Materiales Moleculares, Departamento de Física, Universidad de La Laguna, 38201 Tenerife, Spain
First published on 14th September 2020
Abstract
Thermal-assisted spin crossover and field-induced slow magnetic relaxation coexist in the solid state for the mononuclear cobalt(II) complex with the non-innocent 2,6-bis(N-4-methoxyphenylformimidoyl)pyridine ligand. One-electron oxidation of the paramagnetic low-spin CoII ion (SCo = 1/2) to the diamagnetic low-spin CoIII ion (SCo = 0) leads to the electroswitching of the slow magnetic relaxation in acetonitrile solution.
Metal complexes set up a promising way for the miniaturisation of electronic circuits leading to highly integrated electron spin-based systems for molecular spintronics and quantum computation.1 Combining many molecular magnetic components with multiresponsive and multifunctional abilities is necessary to build up molecular spintronic integrated circuits.2–8 In particular, redox-active mononuclear complexes exhibiting spin crossover (SCO) or single-molecule magnet (SMM) behaviour are of potential interest as electro-switchable molecular spintronic devices (spin transistors, rectifiers, and valves) and quantum devices (qubits and quantum gates).9–30
The combination of SCO and redox properties in one single molecule is allowed in the well-known bis-chelating cobalt(II) complexes with functionalised pyridine-2,6-diimine (PDI) ligands.31–34 The reduction of non-innocent PDI derivatives adds extra electronic features to the oxidizable metal centre in their complexes becoming molecular switches. Here we report the synthesis, structural and spectroscopic characterisation, electrochemical and magnetic properties of a novel mononuclear cobalt(II) complex of formula [Co(4-MeOPhPDI)2](ClO4)2 (1) [4-MeOPhPDI = 2,6-bis(N-4-methoxyphenylformimidoyl)pyridine] and its corresponding singly oxidised cobalt(III) derivative of formula [Co(4-MeOPhPDI)2](Br3)2.35(Br5)0.65·1/4H2O (2). To our knowledge, 1 is the first member of a new class of electroactive molecular nanomagnets that combine both SCO and SMM properties.
4-MeOPhPDI was synthesised from the straightforward condensation of pyridine-2,6-diformaldehyde and 4-anisidine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) in diluted acetic acid media, as reported earlier for the parent unsubstituted PhPDI ligand.32 The reaction between cobalt(II) perchlorate hexahydrate and 4-MeOPhPDI (1:2 molar ratio) in methanol led to 1. Subsequent chemical oxidation of 1 with an excess of bromine in acetonitrile resulted in 2. X-ray suitable single crystals of 1 and 2 were grown in a test tube by layering ethyl acetate on acetonitrile solutions (Fig. S1, ESI‡).
:2 molar ratio) in diluted acetic acid media, as reported earlier for the parent unsubstituted PhPDI ligand.32 The reaction between cobalt(II) perchlorate hexahydrate and 4-MeOPhPDI (1:2 molar ratio) in methanol led to 1. Subsequent chemical oxidation of 1 with an excess of bromine in acetonitrile resulted in 2. X-ray suitable single crystals of 1 and 2 were grown in a test tube by layering ethyl acetate on acetonitrile solutions (Fig. S1, ESI‡).
1 and 2 crystallise in the P43 and I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m space groups of the tetragonal system, respectively (Table S1, ESI‡). The structures of 1 and 2 consist of mononuclear [CoII(4-MeOPhPDI)2]2+ (1) and [CoIII(4-MeOPhPDI)2]3+ (2) complex cations (Fig. 1), perchlorate (1) or tri- and pentabromide (2) anions, and crystallisation water molecules (2). Unlike in 2, the mononuclear complex cations in 1 interact each other through relatively weak intermolecular “face-to-face” π–π stacking interactions between the terminal p-methoxy-substituted phenyl rings (h = 3.86–4.45 Å; θ = 16.3–26.8°) (Fig. S2 and S3, ESI‡). The values of the shortest intermetallic distance through the “face-to-face” π-stacked units in 1 cover the range 9.359(1)–9.795(1) Å, while they range from 9.698(2) to 11.193(2) Å in 2.
2m space groups of the tetragonal system, respectively (Table S1, ESI‡). The structures of 1 and 2 consist of mononuclear [CoII(4-MeOPhPDI)2]2+ (1) and [CoIII(4-MeOPhPDI)2]3+ (2) complex cations (Fig. 1), perchlorate (1) or tri- and pentabromide (2) anions, and crystallisation water molecules (2). Unlike in 2, the mononuclear complex cations in 1 interact each other through relatively weak intermolecular “face-to-face” π–π stacking interactions between the terminal p-methoxy-substituted phenyl rings (h = 3.86–4.45 Å; θ = 16.3–26.8°) (Fig. S2 and S3, ESI‡). The values of the shortest intermetallic distance through the “face-to-face” π-stacked units in 1 cover the range 9.359(1)–9.795(1) Å, while they range from 9.698(2) to 11.193(2) Å in 2.
 | ||
| Fig. 1 Perspective views of the mononuclear cobalt(II) and cobalt(III) cationic units of 1 (a) and 2 (b), respectively, with the atom numbering scheme for the first coordination sphere of the metal atoms [symmetry operation: (I) = −x, y, −z]. The intramolecular π–π stacking interactions are drawn as dotted lines (each of the two ligand backbones are drawn in light and dark grey colours for clarity). | ||
The four crystallographically independent mononuclear cobalt(II) units of 1 exhibit an approximate C2 molecular symmetry (Fig. 1a and Fig. S2, ESI‡). Instead, 2 has a unique crystallographically independent mononuclear cobalt(III) unit with a crystallographically imposed C2 molecular symmetry (Fig. 1b). The occurrence of moderate to strong, intramolecular “face-to-face” π–π stacking interactions between the central pyridine and terminal phenyl rings of different ligands causes a distortion from orthogonality at the Co1 and Co3 atoms in 1 (h = 3.84 and 3.94 Å and θ = 23.5 and 23.4°; Table S2, ESI‡) and the Co1 atom in 2 (h = 3.91 Å and θ = 22.9°; Table S2, ESI‡). The value of the dihedral angle between the mean planes of the diiminomethylpyridine fragments in 2 (Φ = 80.8°) is intermediate between those found in 1 [Φ = 72.2 (Co1), 72.7 (Co3), and 89.9° (Co2 and Co4)].
All cobalt atoms in both 1 and 2 exhibit an axially compressed octahedral geometry. The equatorial plane is defined by the two pairs of imine nitrogen atoms (Nim and Nim′) from each ligand whereas the pyridine nitrogen (Npy) atoms fill the axial positions (Fig. 1). Overall, the metal to nitrogen bond lengths in 1 and 2 are typical of low-spin (LS) CoII and CoIII ions, respectively. The average values of the axial Co–Npy distances are notably shorter than those of the equatorial Co–Nim ones for both 1 [Rax = 1.872(5)–1.940(5) Å and Req = 2.082(4)–2.146(4) Å] and 2 [Rax = 1.841(8) Å and Req = 1.982(9) Å].
The χMT vs. T plot (χM being the dc molar magnetic susceptibility per mononuclear unit and T the absolute temperature) for 1 reveals the presence of a gradual but complete, thermally-induced SCO (Fig. 2), as reported earlier for the parent cobalt(II) complex with the unsubstituted PhPDI ligand.32 In both cases, no hysteresis occurs despite the occurrence of intermolecular π–π stacking interactions in the solid state. The χMT value of 1.42 cm3 mol−1 K at room temperature is lower than that expected for a high-spin (HS) CoII ion with an unquenched orbital momentum contribution (χMT = 2.70 cm3 mol−1 K with SCo = 3/2 and gCo = 2.4). Upon cooling, χMT decreases smoothly to reach a sort of plateau around 100 K with a χMT value of 0.45 cm3 mol−1 K at 2.0 K, as expected for a LS CoII ion (χMT = 0.45 cm3 mol−1 K with SCo = 1/2 and gCo = 2.2).
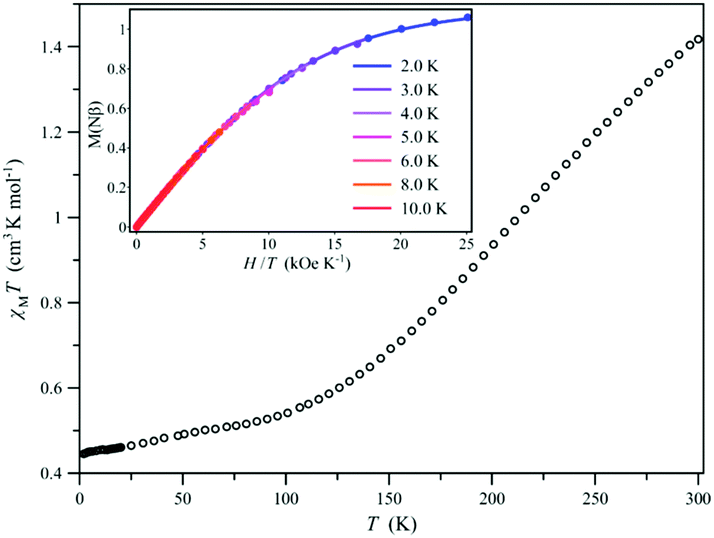 | ||
| Fig. 2 Temperature dependence of χMT for 1 under applied dc magnetic fields of 0.25 (T ≤ 20 K) and 5.0 kOe (T > 20 K). The inset shows the field dependence of M for 1 in the temperature range of 2.0–10.0 K. The solid line is the best-fit curve (see text). | ||
This conclusion is also supported by the M vs. H/T plots (M being the molar magnetisation per mononuclear unit and H the applied dc magnetic field). Hence, they show: (i) a maximum value of M about 1.00 Nβ at H = 50 kOe and T = 2.0 K close to that for a LS CoII ion (M = 1.10 Nβ with gCo = 2.2); (ii) the superimposition of the reduced isothermal magnetisation curves in the temperature range of 2.0–10.0 K; and (iii) the good simulation of these curves by the Brillouin function with SCo = 1/2 and gCo = 2.20 (inset of Fig. 2). On the other hand, the X-band EPR spectrum of 1 at 4.0 K in the solid state (Fig. 3) exhibits an axial signal (gx = gy = 2.09 and gz = 2.42), which is similar to that in frozen acetonitrile solutions (gx = gy = 2.08 and gz = 2.37). This situation is typical of a LS CoII ion with a dx2−y2 magnetic orbital, as expected for an axially compressed coordination geometry, both in the solid state and in solution.
 | ||
| Fig. 3 X-band EPR spectra of 1 at 4.0 K, both in the solid state (pale green line) and in a frozen acetonitrile solution (dark green line). The inset shows the X-band EPR spectra of 1 and 2 at 4.0 K in frozen acetonitrile solution (green and red solid lines), compared with those of the corresponding in situ monoxidised and monoreduced species (red and green dotted lines). | ||
The χM′ and χM′′ vs. ν plots of 1 (χM′ and χM′′ being the in-phase and out-of-phase ac molar magnetic susceptibilities per mononuclear unit) are characteristic of field-induced SMMs, both in the solid state and in frozen acetonitrile solutions. In this regard, there are two related examples of square-pyramidal mononuclear SCO cobalt(II) complexes exhibiting a field-induced SMM behaviour in the solid state,35,36 which have been proposed as promising candidates to molecular electron spin-based quantum bits (qubits).37
Neither frequency dependence of χM′ nor a χM′′ signal are observed in the absence of a dc magnetic field (data not shown). Some authors usually explain these features by a rapid relaxing (RR) spin featuring a fast quantum tunnelling of magnetisation (QTM).35,36 However, this phenomenon requires a barrier, which is not possible for a doublet spin ground state, as in 1. Frequency-dependent χM′ and χM′′ signals arise by applying a magnetic field (Hdc = 1.0, 2.5 and 5.0 kOe in solid state and 2.5 kOe in solution) in agreement with the occurrence of a field-induced slow relaxing (SR) LS CoII ion (Fig. S5 and S6, ESI‡). Hence, incipient signals develop at the χM′′ vs. ν plots with decreasing temperature, both in the solid state and in solution. Additionally, in both conditions, a distinct maximum appears at the χM′′ vs. ν plots that progressively shifts toward lower frequencies of the oscillating ac magnetic field as temperature decreases. This maximum occurs even at a higher temperature in solution. Consequently, the field-induced SR behaviour is not only retained but enhanced in frozen acetonitrile solutions relative to the solid state.
The values of the magnetic relaxation time (τ) were calculated from the joint analysis of the χM′ and χM′′ vs. ν plots through the generalised Debye equations, which consider the static (χS) and infinite (χT) frequency magnetic susceptibilities, and the exponential factor that describes the broadness of the spectra (α), as additional fitting parameters (Fig. S7, ESI‡). The results displayed as lnτ vs. 1/T graphs (Arrhenius plots) were satisfactorily simulated considering a simple magnetic two-phonon Raman relaxation mechanism, τ−1 = CTn (Fig. 4). However, in the range of lower temperatures, at 5.0 kOe in solid state and 2.5 kOe in solution, required the inclusion of an extra relaxation process. Unexpectedly, this additional process was not a one-phonon direct mechanism,35,36 but a temperature-independent term (τ−1 = k). This contribution to the spin-reversal relaxation should occur within the Zeeman-split mS = ±1/2 Kramers ground doublet and should be seen as a zero-order term of a direct mechanism and not as a QTM process inconsistent with an S = 1/2 spin ground state. This Hdc dependent component should be notable in the lack of an applied magnetic field and, therefore, responsible for the zero-field RR behaviour.
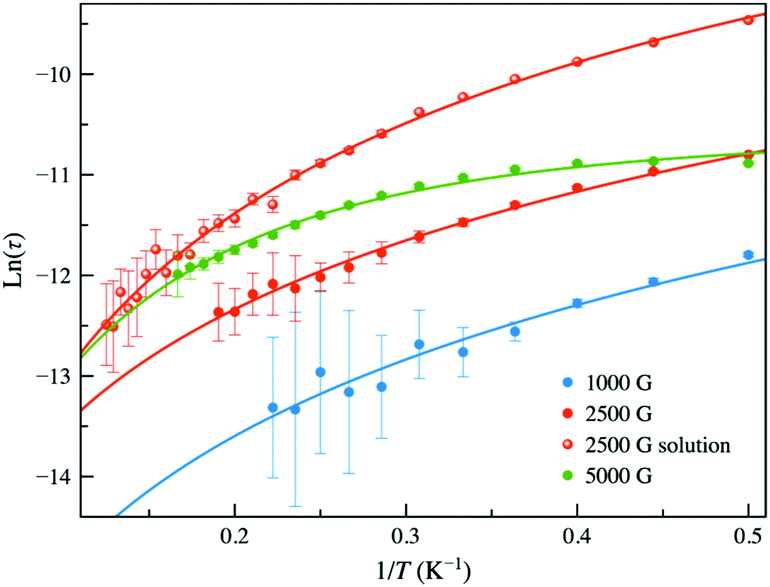 | ||
| Fig. 4 Arrhenius plots of 1 under applied dc magnetic fields of 1.0, 2.5 and 5.0 kOe in the solid state and 2.5 kOe in solution. The solid lines are the best-fit for a double relaxation mechanism described by the equation τ−1 = CTn + k (see text). Vertical error bars denote the standard deviations. | ||
The calculated values of the coefficients of the τ−1 = CTn + k model appear summarised in Table S3 (ESI‡). The low values of n (1.87–2.45) justify the needless inclusion of a first-order direct relaxation term (τ−1 = AT) and suggest the presence of optical rather than acoustic phonons. For the Raman mechanism, the similarity between the results in solution and the solid state at a larger magnetic field could lie in a decrease of the effects of the network when Hdc is increased.
The cyclic voltammograms of 1 in acetonitrile at room temperature show one reversible oxidation wave at E1 = +0.39 V vs. the ferricinium/ferrocene (Fc+/Fc) couple, together with up to four reduction waves at E2 = −0.74 V, E3 = −1.62 V, E4 = −2.02 V, and E5 = −2.19 V, where only the two first ones are reversible (Fig. 5). This conclusion is supported by: (i) the values of the anodic to cathodic peak separation of these three reversible redox waves (ΔEp,1 = 83 mV, ΔEp,2 = 81 mV and ΔEp,3 = 77 mV), which are comparable to that of the Fc+/Fc couple (ΔEp = 80 mV) under the same conditions (inset of Fig. 5), and (ii) the linearity of the peak current against the square root of the scan rate plots for each of them (data not shown).
 | ||
| Fig. 5 Cyclic voltammograms of 1 in acetonitrile (0.1 M nBu4NPF6) at 25 °C and 200 mV s−1. The inset shows the cyclic voltammograms of the reversible redox waves in the scan rate range of 20–250 mV s−1. | ||
The assignment of the observed redox processes for 1 is illustrated in Scheme S1 (ESI‡).34 The reversible oxidation wave corresponds to the one-electron metal oxidation to afford the cobalt(III) species, while the first pair of reversible reduction waves conforms to the stepwise one-electron reduction of the ligands to give the single and double imide-type π-radical anion cobalt(II) species. That being so, the second pair of irreversible reduction waves is likely attributed to the further stepwise one-electron reduction of the ligands to give the single and double imide-type dianion cobalt(II) species. Both the σ-donor capacity of the electron-donating methoxy ligand substituents at the terminal phenyl groups and the π-acceptor ability of the central pyridinediimine ligand fragment are the leading cause of this dual metal- and ligand-centred multiredox behaviour of 1.
The reversible interconvertion between the paramagnetic cobalt(II) complex and the diamagnetic cobalt(III) derivative has been monitored through X-band EPR spectra in solution (inset of Fig. 3). Upon addition of an excess of bromine to an acetonitrile solution of 1, the axial EPR signal of the paramagnetic LS CoII ion (SCo = 1/2) disappears to give the corresponding EPR-silent diamagnetic LS CoIII (SCo = 0) oxidised species. Conversely, the axial EPR signal of 1 is restored by addition of a stoichiometric amount of hydrazine to an acetonitrile solution of 2.
A solution of 1 in acetonitrile at 0 °C was subjected to electrochemical oxidation (Eox = +1 V) and subsequent reduction (Ered = 0 V) (Fig. S4, ESI‡). Notably, the slow magnetic relaxation is preserved, the χM′ and χM′′ vs. T plots before and after the electrolysis cycle being almost identical (Fig. S8, ESI‡). In both cases, distinct χM′ and χM′′ maxima appear and progressively shift toward higher temperatures as far as the frequency of the oscillating ac magnetic field increases. This reversible response demonstrates that this interconvertible system works as a genuine molecular magnetic electroswitch in solution, even if it proceeds as two different compounds in the solid state.
In summary, we report a unique example of mononuclear cobalt(II) complex with a non-innocent pyridinediimine (PDI) ligand that exhibits both thermal-assisted LS–HS and field-induced RR–SR transitions. Moreover, it shows a dual redox behaviour featuring one-electron oxidation of the slow-relaxing paramagnetic LS CoII ion (SCo = 1/2) to the diamagnetic LS CoIII ion (SCo = 0) and stepwise two-electron reduction of the PDI ligands to the corresponding imide-type PDI˙− π-radical anion and PDI2− dianion. This new class of triply thermal-, magneto-, and electro-switchable molecular nanomagnet possesses multiple metal- or ligand-based, redox and spin states, which reversibly interconvert under different external stimuli (temperature, magnetic field, and redox potential). The reported magnetic and electronic multistability in this mononuclear cobalt(II)–PDI complex illustrates the potential of multiresponsive and multifunctional SCO/SMM molecules in the emerging fields of molecular spintronics and quantum computation.
This work was supported by the Spanish MINECO (Projects PID2019-109735GB-I00, CTQ2016-75068P, MAT2017-89203-R, and Unidad de Excelencia María de Maeztu MDM-2015-0538). This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 748921-SIMOF. R. R. and J. P. thank the Generalitat Valenciana and the Cabildo de Tenerife for a doctoral grant (GRISOLIAP/2017/057) and the Agustín de Betancourt research contract, respectively. We are indebted to Drs Marta Viciano-Chumillas and Gloria Agustí (Universitat de València) for the assistance in the electrochemical and EPR spectroscopic measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martínez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-García and M. Julve, Coord. Chem. Rev., 2017, 339, 17 CrossRef CAS.
- A. Dei, D. Gatteschi, C. Sangregorio and L. Sorace, Acc. Chem. Res., 2004, 37, 827 CrossRef CAS.
- O. Sato, J. Tao and E.-J. Zhang, Angew. Chem. Int. Ed., 2007, 46, 2152 CrossRef CAS.
- M. Castellano, R. Ruiz-García, J. Cano, J. Ferrando-Soria, E. Pardo, F. R. Fortea-Pérez, S. Eddine-Stiriba, M. Julve and F. Lloret, Acc. Chem. Res., 2015, 48, 510 CrossRef CAS.
- B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250 CrossRef CAS.
- D. Aguilà, Y. Prado, E. S. Koumousi, C. Mathonière and R. Clérac, Chem. Soc. Rev., 2016, 45, 203 RSC.
- M. M. Khusniyarov, Chem. – Eur. J., 2016, 22, 15178 CrossRef CAS; B. Doistau, L. Benda, B. Hasenknopf, V. Marvaud and G. Vives, Magnetochemistry, 2018, 4, 1 Search PubMed.
- Y.-S. Meng and T. Liu, Acc. Chem. Res., 2019, 52, 1369 CAS.
- J. J. Parks, A. R. Champagne, T. A. Costi, W. W. Shum, A. N. Pasupathy, E. Neuscamman, S. Flores-Torres, P. S. Cornaglia, A. A. Aligia, C. A. Balseiro, G.-K.-L. Chan, H. D. Abruña and D. C. Ralph, Science, 2010, 328, 1370 CrossRef CAS.
- V. Meded, A. Bagrets, K. Fink, R. Chandrasekar, M. Ruben, F. Evers, A. Bernand-Mantel, J. S. Seldenthuis, A. Beukman and H. S. J. van der Zant, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245415 CrossRef.
- T. Mahfoud, G. Molnár, S. Cobo, L. Salmon, C. Thibault, C. Vieu, P. Demont and A. Bouseksou, Appl. Phys. Lett., 2011, 99, 053307 CrossRef.
- P. Verma, J. Weir, L. Mirica and T. D. Stack, Inorg. Chem., 2011, 50, 9816 CrossRef CAS; D. Aravena and E. Ruiz, J. Am. Chem. Soc., 2012, 134, 777 CrossRef.
- M. G. Cowan, J. Olguín, S. Narayanaswamy, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2012, 134, 2892 CrossRef CAS.
- E. Ruiz, Phys. Chem. Chem. Phys., 2014, 16, 14 RSC.
- G. D. Harzmann, R. Frisenda, H. S. J. van der Zant and M. Mayor, Angew. Chem. Int. Ed., 2015, 54, 13425 CrossRef CAS.
- E. J. Devid, P. N. Martinho, M. V. Kamalakar, I. Salitros, U. Prendergast, J.-F. Dayen, V. Meded, T. Lemma, R. González-Prieto, F. Evers, T. E. Keyes, M. Ruben, B. Doudin and S. J. van der Molen, ACS Nano, 2015, 9, 4496 CrossRef CAS.
- A. C. Aragonès, D. Aravena, J. I. Cerdá, Z. Acís-Castillo, H. Li, J. A. Real, F. Sanz, J. Hihath, E. Ruiz and I. Díez-Pérez, Nano Lett., 2016, 16, 218 CrossRef.
- A. C. Aragonès, D. Aravena, F. J. Valverde-Muñoz, J. A. Real, F. Sanz, J. Hihath, I. Díez-Pérez and E. Ruiz, J. Am. Chem. Soc., 2017, 139, 5768 CrossRef.
- T. Jasper-Toennies, M. Gruber, S. Karan, H. Jacob, F. Tuczek and R. Berndt, Nano Lett., 2017, 17, 6613 CrossRef CAS.
- L. D. Wickramasinghe, S. Mazumder, K. K. Kpogo, R. J. Staples, H. B. Schlegel and C. N. Verani, Chem. – Eur. J., 2016, 22, 10876 CrossRef.
- N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498 CrossRef CAS.
- S. Takamatsu, T. Ishikawa, S.-Y. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250 CrossRef CAS.
- S. Kyatskaya, J. R. Galán-Mascarós, L. Bogani, F. Hennrich, M. Kappes, W. Wernsdorfer and M. Ruben, J. Am. Chem. Soc., 2009, 131, 15143 CrossRef CAS.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef CAS.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357 CrossRef CAS.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 165 CrossRef CAS.
- E. M. Fatila, M. Roùzieres, M. C. Jennings, A. J. Lough, R. Clérac and K. E. Preuss, J. Am. Chem. Soc., 2013, 135, 9596 CrossRef CAS.
- L. Norel, M. Feng, K. Bernot, T. Roisnel, T. Guizouarn, K. Costuas and S. Rigaut, Inorg. Chem., 2014, 53, 2361 CrossRef CAS.
- M. Damjanovic, T. Morita, K. Katoh, M. Yamashita and M. Enders, Chem. – Eur. J., 2015, 21, 14421 CrossRef CAS.
- M. A. Antunes, J. T. Coutinho, I. C. Santos, J. Marçalo, M. Almeida, J. J. Baldoví, L. C. J. Pereira, A. Gaita-Ariño and E. Coronado, Chem. – Eur. J., 2015, 21, 17817 CrossRef CAS.
- P. E. Figgins and D. H. Busch, J. Am. Chem. Soc., 1960, 82, 820 CrossRef CAS.
- G. Juhász, S. Hayami, K. Inoue and Y. Maeda, Chem. Lett., 2003, 32, 882 CrossRef.
- B. De Bruin, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 39, 2936 CrossRef CAS.
- C. Römelt, T. Weyhermüller and K. Wieghardt, Coord. Chem. Rev., 2019, 380, 287 CrossRef.
- H.-H. Cui, J. Wang, X.-T. Chen and Z.-L. Xue, Chem. Commun., 2017, 53, 930 Search PubMed.
- L. Chen, J. Song, W. Zhao, G. Yi, Z. Zhou, A. Yuan, Y. Song, Z. Wang and Z.-W. Ouyang, Dalton Trans., 2018, 47, 16596 RSC.
- M.-X. Xu, Z. Liu, B.-W. Dong, H.-H. Cui, Y.-X. Wang, J. Su, Z. Wang, Y. Song, X.-T. Chen, S.-D. Jiang and S. Gao, Inorg. Chem., 2019, 58, 2330 CrossRef CAS.
Footnotes |
| † To the memory of Prof. Catalina Ruiz-Pérez, outstanding researcher and excellent teacher, best friend and colleague who passed away in August 2019. |
| ‡ Electronic supplementary information (ESI) available: Experimental details on the synthesis of the ligand and complexes and their physicochemical characterisation (Fig. S1–S8, Scheme S1, and Tables S1–S3). CCDC 1989324 and 1989325. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03357j |
| This journal is © The Royal Society of Chemistry 2020 |