
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Step and redox efficient nitroarene to indole synthesis†
Bünyamin
Özkaya
 ,
Christina L.
Bub
and
Frederic W.
Patureau
*
,
Christina L.
Bub
and
Frederic W.
Patureau
*
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: Frederic.patureau@rwth-aachen.de; Web: https://www.patureau-oc-rwth-aachen.de
First published on 6th October 2020
Abstract
Step and redox efficiencies are a rising priority in synthetic method development, in order to make synthetic processes more sustainable and more affordable. Herein, a step and redox efficient nitroarene to indole synthesis is developed, in sharp contrast to the rich literature on the construction of indoles. Elemental zinc was found to be the best terminal reductant.
The indole scaffold is one of the most important heterocyclic motifs in organic chemistry. Its frequent occurrence in pharmaceuticals and natural products and its biological properties make it an essential basic structure.1–5 This has led to ongoing interest in the scientific community on synthesizing indoles using different approaches, leading to many strategies.6–13 One of the earliest is of course the Fischer indole synthesis, arguably a remarkably powerful coupling reaction.1,14,15
Much of early organic chemistry, however, focused on fine-tuning the reacting functional groups, often through multiple (redox) pre-activation steps, in order to trick the desired coupling reactions into occurring (Fig. 1). The Fischer indole synthesis is a good example of this problem (Scheme 1a). Indeed, the starting arene substrate on which one wishes to build an indole must be oxidized and reduced back and forth a few times in order to reach the desired hydrazine reactive state. Only then will the Fischer indole coupling occur in a final overall redox neutral step through condensation with ketones. Meanwhile, the development of more sustainable synthetic methods has become a major concern and priority in order to reduce the environmental footprint of these desired processes. In principle, this can be achieved by reducing the number of superfluous redox operations, and thereby the associated chemical waste. In 1991, Larock utilized the recent progress made in Pd-catalysed cross-coupling chemistry to propose a shorter route towards the indole scaffold, through pre-activation of the ortho position of the aniline intermediate with a halide.16–19 This is then followed by a classical cross-coupling step with internal alkynes (i.e., dehydrated ketones) under Pd-catalysis (Scheme 1a).
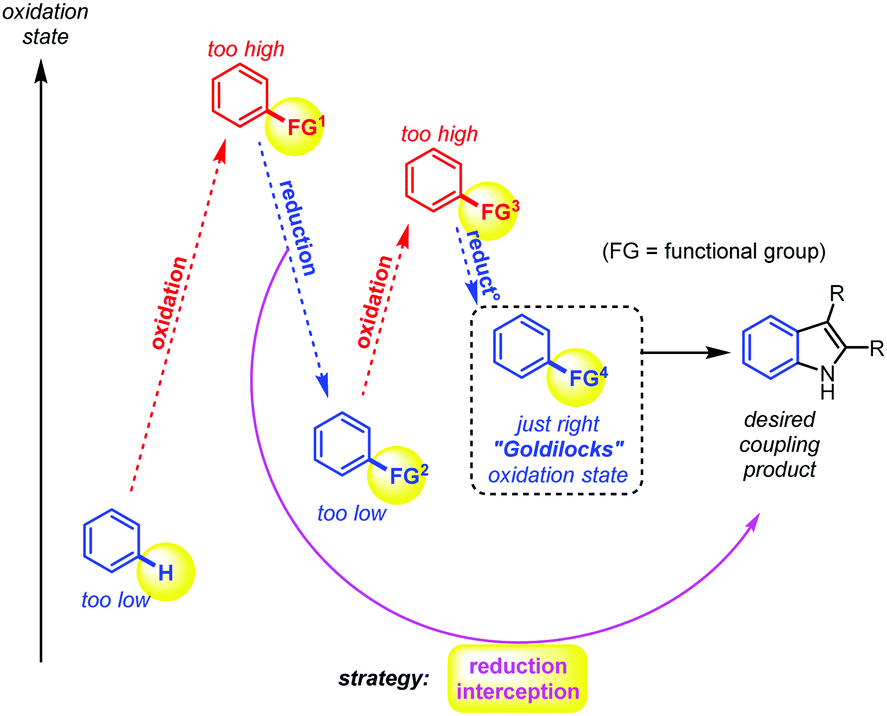 | ||
| Fig. 1 The problem of step and redox efficiency in synthetic methodology: fine-tuning the functional groups for a desired coupling reaction. | ||
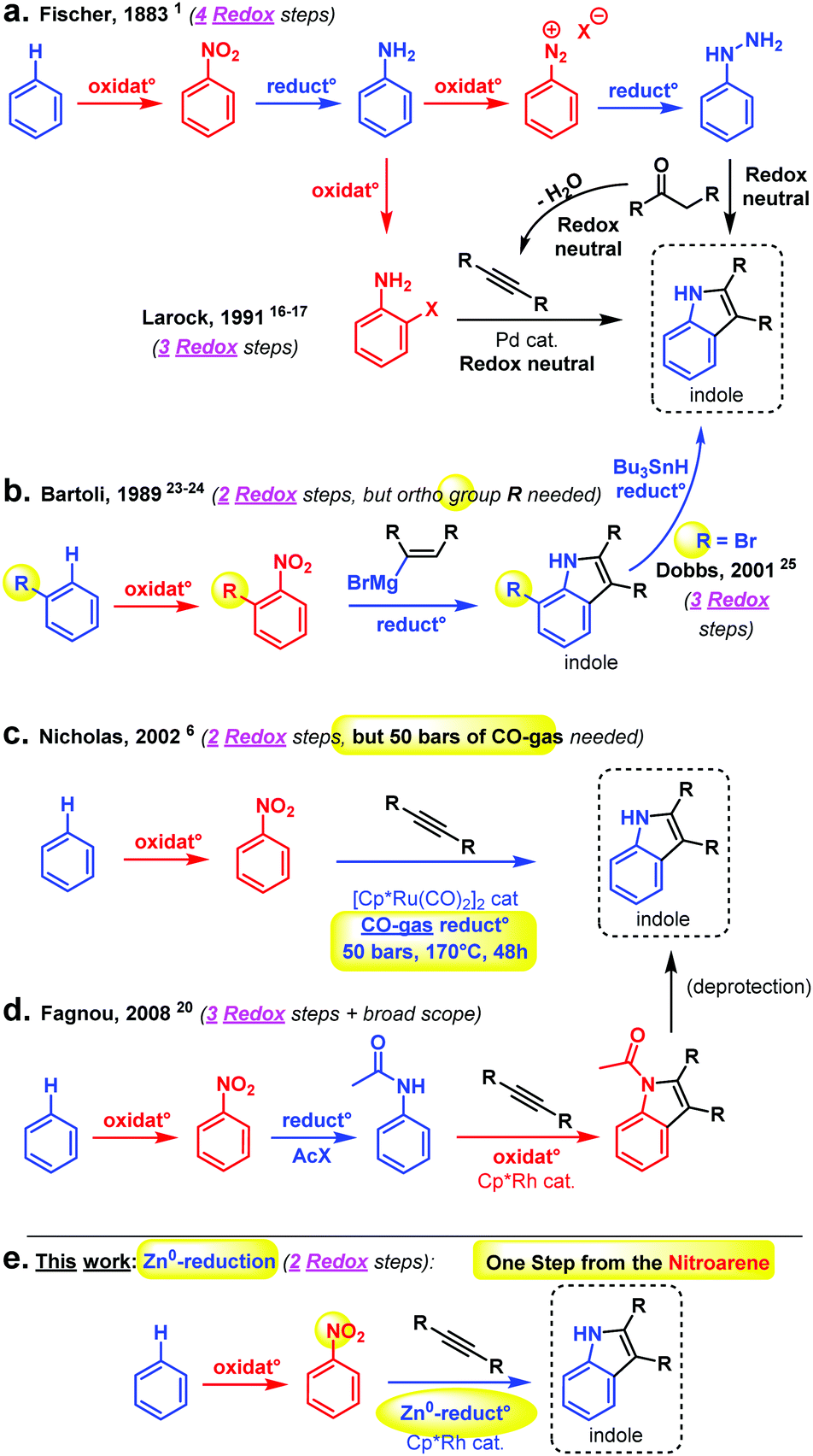 | ||
| Scheme 1 (a) Fischer indole synthesis (4 redox steps) and indole synthesis by Larock using Pd-catalysis and a pre-functionalized aniline (3 redox steps), (b) Bartoli synthesis requiring ortho-substituted nitrobenzenes (2 redox steps), and Dobbs synthesis utilizing 1,2-bromonitroarenes, then removing the halide with Bu3SnH (3 redox steps), (c) Nicholas Ru-catalysed indole synthesis with 50 bars of CO gas as the reductant, (d) Rh-catalysed indole synthesis starting from acetanilides (3 redox steps), and (e) this work: reductive nitroarene to indole synthesis (directly from the nitroarene) with Zn0 powder. | ||
In 2008, Fagnou, inspired by the then recent emergence of Rh-catalysed C–H bond activation, improved this alkyne approach further utilizing the corresponding protected acetanilide under oxidative conditions.20–22 While the scope of that reaction is generally broad, making it an important synthetic tool, the Fagnou approach represents still three redox operations from the arene starting material: (1) (oxidative) nitration, (2) reduction to the aniline or anilide, and (3) oxidative Rh-catalysed coupling with the alkyne. Moreover, getting to the target indole requires a final deacetylation/deprotection step. In terms of the overall step and redox efficiency, at least with respect to the arene substrate, one of the most straightforward approaches remains that of Bartoli with his direct synthesis of indoles from nitroarenes and vinyl organomagnesium reagents (1989).23,24 Bartoli's strategy features only two redox operations: (1) (oxidative) nitration, and (2) reductive coupling with an organometallic species, which is both nucleophile and reductant (Scheme 1b).
Unfortunately, Bartoli's indole synthesis is associated with two main limitations: (1) the harshness of the organomagnesium reagent, thus limiting functional group tolerance and, more problematically: (2) the requirement for an ortho-functional group at the nitroarene substrate for the coupling reaction to occur. In 2001, Dobbs found one way around the latter problem by utilizing 1,2-bromonitroarenes, thereafter removing the ortho bromide in a third redox operation with Bu3SnH as a – not so convenient nor environmentally friendly – final reductant.25 One of the most step and redox efficient indole syntheses in the literature is probably that of Nicholas (2002, Scheme 1c).6 This early and inspiring Ru-catalysed C–H bond activation reaction requires, however, as much as 50 bars of toxic CO gas as the reductant, 170 °C reaction temperature, and 48 h reaction time for only very moderate yields of indole coupling products. Several other methods exist for the construction of the indole scaffold, though usually these do not operate directly from the nitroarene but rather from a derivative of it, and involve therefore at least one additional redox operation in the retrosynthetic strategy.26–28 For the sake of clarity, not all methods could be graphically summarized in Scheme 2. Thus, clearly, practical step and redox efficient strategies to build the indole scaffold are still in high demand, in particular using non-ortho functionalized nitroarenes. Herein, we propose to develop such a strategy, by directly intercepting the reduction of the nitroarene substrate29–46 to achieve a straightforward Rh-catalysed C–H bond activation47–52 event and coupling with the alkyne.
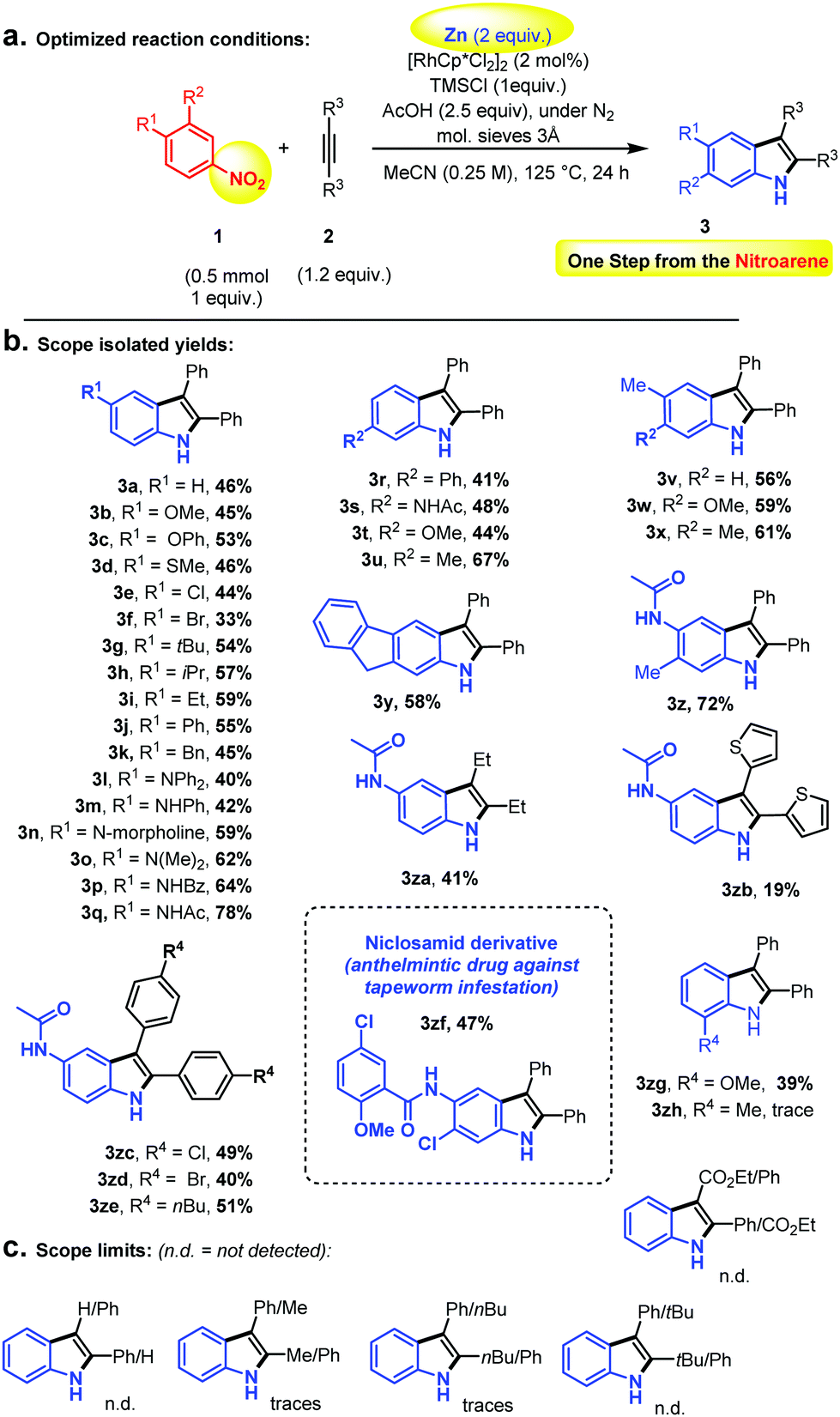 | ||
| Scheme 2 (a) Optimized reaction conditions, (b) scope, isolated yields, and (c) scope limits. | ||
We thus started with nitrobenzene and diphenylacetylene as model substrates, in the presence of catalytic amounts of the classical [Cp*RhCl2]2 dimer.47–52 The fairly simple idea was that although the nitro group is not reputed to be an efficient C–H bond activation ortho directing group, some of its reduction intermediates (i.e., nitroso or similar)53 – if sufficiently long-lived – might be more susceptible. Of course, the challenge was to achieve Rh-catalysed ortho C–H bond activation and functionalization before full reduction to the aniline, at which point any directing group and C–H oxidative ability would have been suppressed. Indeed, primary anilines are not reputed as efficient C–H bond activation ortho directing groups either. Large numbers of additives and especially reductants were therefore screened, as well as temperature and solvent. The full optimization table is given in the ESI.† Eventually, elemental Zn-powder was found to be the optimal reductant, in the presence of TMSCl, acetic acid and 3 Å molecular sieves, in acetonitrile. Full conversion to the corresponding product was never observed, due to the formation of homo-coupled over-reduction by-products (i.e., hydrazines). Moreover, relatively long reactions times were found to be necessary (very little product formation in the first hours, 24 h reaction time recommended), thus highlighting the kinetic challenge of performing nitro reduction concomitantly to ortho directed C–H bond activation/functionalization. Importantly, the indole coupling product is not observed unless the solvent is a nitrile. Utilizing a RhCp* source turned out to be crucial, since other Rh-catalysts did not give any desired product. Organic reductants or other metal salts turned out to be inefficient in the reaction. Moreover, replacing TMSCl with TMSI or a different Si-species gave only trace amounts or no product at all. The optimized conditions and the substrate scope are depicted in Scheme 2. Overall, the isolated yields were found promising. Ethers (3b and 3c), methylthioether (3d) and halides (3e and 3f) in the para-position with respect to the nitro-group gave moderate yields. Alkyl and aryl groups (3g–k, 3u–v, 3x–y) as well as different kinds of amines/amides (3l–q) led smoothly to the corresponding coupling products.
Moreover, meta functionalization of the nitroarene substrates led to outstanding C–H bond functionalization regio-selectivity (3r–za), a phenomenon that was already observed by Fagnou and co-authors under oxidative reaction conditions.20 Indeed, the other regioisomers (C–H bond functionalization on the more sterically congested ortho position) could not be detected. This outstanding selectivity can be interpreted as the result of a sterically controlled highly regioselective alkyne insertion into the C–Rh bond subsequent to the C–H bond activation event. This high sensitivity to steric effects may also explain the more moderate performance of ortho functionalized nitroarenes (3zg–zh), therefore highlighting a useful methodological orthogonality with the Bartoli reaction. It is unclear however why unsymmetrically substituted alkynes should fail (Scheme 2) in this reaction. Nevertheless, surprisingly, the anilide functional group (3p–q, 3z) is particularly well tolerated. This is interesting because anilides are privileged directing groups in the field of C–H bond activation,20 thus offering further functionalization possibilities. In addition, this has allowed the late stage C–H “indolisation” of an important anthelmintic drug, Niclosamid, with a promising 47% isolated yield (3zf) and excellent regioselectivity (Scheme 2b).
In order to gain some insights into the role and oxidation states of the directing group, some selected control experiments were conducted (Scheme 3). Of course, as the coupling reaction was optimized for trivial nitroarenes, it is not too surprising to find that they were also the best directing group (DG) precursor under those conditions. It is nevertheless interesting that, without altering the reaction conditions, nitrosobenzene 4 and N-phenylhydroxylamine 5 deliver some small isolable amounts of target indole 3a (14% and 5% respectively). However, aniline 6, 1,2-diphenylhydrazine 7 and azobenzene 8 are not competent coupling reagents/directing groups. Moreover, interestingly, when we performed the reaction on nitrosobenzene 4 and N-phenylhydroxylamine 5 in the absence of the zinc terminal reductant, an undefined and complex mixture of products and perhaps some decomposition were observed (Scheme 3). This surprising result – one might have expected a high yield of indole coupling product, or its oxide – contradicts our original simplified mechanistic scenario, wherein (1) reduction of the nitro directing group and (2) C–H activation/insertion/reductive elimination were thought to be sequential and unrelated. In contrast, our results suggest that Zn is more than a simple reductant in this reaction, and might structurally participate in some of the critical steps of the herein described reaction. Intermediate crystallization attempts unfortunately failed so far. Further research efforts are currently ongoing in our laboratory to elucidate and perhaps exploit those effects in other future coupling reactions.
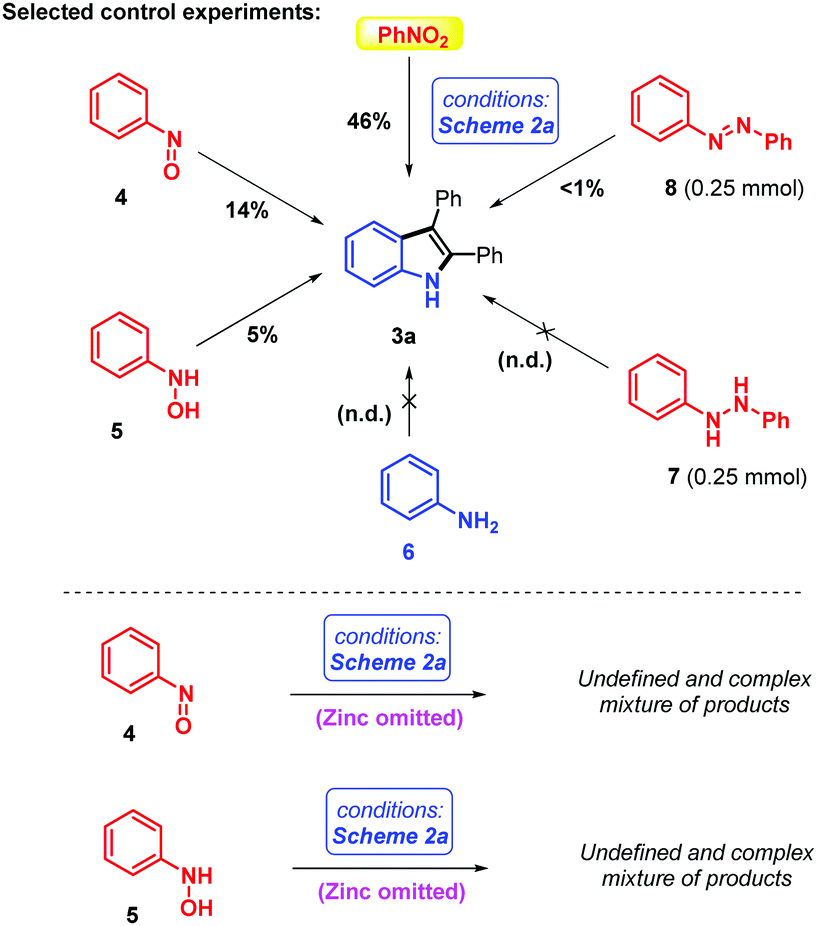 | ||
| Scheme 3 Selected control experiments with different oxidation states of the nitrogen atom under optimized conditions. | ||
In conclusion, we have herein developed a direct nitroarene to indole synthesis, relying on elemental Zn as the terminal reductant. This therefore represents another milestone in the quest for more redox efficient routes towards valuable indole scaffolds. More generally, the relatively underexplored concept of reductive or oxidative process interception might lead to new strategies for the design of innovative coupling reactions.
ERC project 716136:2O2ACTIVATION is acknowledged for generous support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Fischer and O. Hess, Chem. Ber., 1884, 17, 559 CrossRef.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS.
- M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694 RSC.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620 CrossRef CAS.
- J.-H. Lee and J. Lee, FEMS Microbiol. Rev., 2010, 34, 426 CrossRef CAS.
- A. Penoni and K. M. Nicholas, Chem. Commun., 2002, 484 RSC.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS.
- S. Würtz, S. Rakshit, J. J. Neumann, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 7230 CrossRef.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195 CrossRef CAS.
- N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29 RSC.
- T. Guo, F. Huang, L. Yu and Z. Yu, Tetrahedron Lett., 2015, 56, 296 CrossRef CAS.
- S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544 RSC.
- B. Robinson, Chem. Rev., 1963, 63, 373 CrossRef.
- M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7, 52852 RSC.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CrossRef CAS.
- R. C. Larock, E. K. Yum and M. D. Refvik, J. Org. Chem., 1998, 63, 7652 CrossRef CAS.
- L. Ackermann, Organometallics, 2003, 22, 4367 CrossRef CAS.
- M. Shen, G. Li, B. Z. Lu, A. Hossain, F. Roschangar, V. Farina and C. H. Senanayake, Org. Lett., 2004, 6, 4129 CrossRef CAS.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS.
- Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS.
- G. N. Hermann, C. L. Jung and C. Bolm, Green Chem., 2017, 19, 2520 RSC.
- G. Bartoli, G. Palmieri, M. Bosco and R. Dalpozzo, Tetrahedron Lett., 1989, 30, 2129 CrossRef CAS.
- G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728 RSC.
- A. Dobbs, J. Org. Chem., 2001, 66, 638 CrossRef CAS.
- S. Murru, A. A. Gallo and R. S. Srivastava, ACS Catal., 2011, 1, 29 CrossRef CAS.
- S. Murru, A. A. Gallo and R. S. Srivastava, Eur. J. Org. Chem., 2011, 2035 CrossRef CAS.
- G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331 RSC.
- For inspiring prior work with Palladium catalysis and CO gas as the reductant, see for example: F. Ragaini, A. Rapetti, E. Visentin, M. Monzani, A. Caselli and S. Cenini, J. Org. Chem., 2006, 71, 3748 CrossRef CAS.
- J. Liu, C. Zhang, Z. Zhang, X. Wen, X. Dou, J. Wei, X. Qiu, S. Song and N. Jiao, Science, 2020, 367, 281 CrossRef CAS.
- H. Song, Z. Yang, C.-H. Tung and W. Wang, ACS Catal., 2020, 10, 276 CrossRef CAS.
- M. Rauser, R. Eckert, M. Gerbershagen and M. Niggemann, Angew. Chem., Int. Ed., 2019, 58, 6713 CrossRef CAS.
- S. Zhao and N. P. Mankad, Org. Lett., 2019, 21, 10106 CrossRef CAS.
- N. Shen, C. W. Cheung and J.-A. Ma, Chem. Commun., 2019, 55, 13709 RSC.
- L. Ling, C. Chen, M. Luo and X. Zeng, Org. Lett., 2019, 21, 1912 CrossRef CAS.
- C. Lu, Z. Su, D. Jing, S. Jin, L. Xie, L. Li and K. Zheng, Org. Lett., 2019, 21, 1438 CrossRef CAS.
- S. Suárez-Pantiga, R. Hernández-Ruiz, C. Virumbrales, M. R. Pedrosa and R. Sanz, Angew. Chem., Int. Ed., 2019, 58, 2129 CrossRef.
- C. W. Cheung, J.-A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789 CrossRef CAS.
- M. Rauser, C. Ascheberg and M. Niggemann, Chem. – Eur. J., 2018, 24, 3970 CrossRef CAS.
- M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570 CrossRef CAS.
- C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef.
- J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886 CrossRef CAS.
- H. Yan, H. Wang, X. Li, X. Xin, C. Wang and B. Wan, Angew. Chem., Int. Ed., 2015, 54, 10613 CrossRef CAS.
- Z. Zhou, G. Liu, Y. Chen and X. Lu, Adv. Synth. Catal., 2015, 357, 2944 CrossRef CAS.
- C. Wang, D. Wang, H. Yan, H. Wang, B. Pan, X. Xin, X. Li, F. Wu and B. Wan, Angew. Chem., Int. Ed., 2014, 53, 11940 CrossRef CAS.
- L. L. Anderson, M. A. Kroc, T. W. Reidl and J. Son, J. Org. Chem., 2016, 81, 9521 CrossRef CAS.
- F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS.
- J. Park and S. Chang, Chem. – Asian J., 2018, 13, 1089 CrossRef CAS.
- S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304 CrossRef CAS.
- Y. Shang, K. Jonnada, S. L. Yedage, H. Tu, X. Zhang, X. Lou, S. Huang and W. Su, Chem. Commun., 2019, 55, 9547 RSC.
- C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS.
- A. Penoni, J. Volkmann and K. M. Nicholas, Org. Lett., 2002, 4, 699 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization, methods, characterization and spectra. See DOI: 10.1039/d0cc03258a |
| This journal is © The Royal Society of Chemistry 2020 |