
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C(sp3)–H arylation to construct all-syn cyclobutane-based heterobicyclic systems: a novel fragment collection†
Thomas J.
Osberger‡
 ab,
Sarah L.
Kidd‡
a,
Thomas A.
King
a and
David R.
Spring
*a
ab,
Sarah L.
Kidd‡
a,
Thomas A.
King
a and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bDepartment of Chemistry and Biochemistry, California State Polytechnic University, Pomona, CA, USA
First published on 26th May 2020
Abstract
All-syn fused cyclobutanes remain an elusive chemotype and thus present an interesting synthetic challenge. Herein, we report the successful application of Pd-catalysed C(sp3)–H arylation of cyclobutane compounds to generate all-syn heterobicyclic fragments using an innovative ‘inside-out’ approach. Through this strategy we generate a virtual collection of 90 fragments, which we demonstrate to have enhanced three-dimensionality and superior fragment-like properties compared to existing collections.
Fragment-based drug discovery (FBDD) has emerged as a powerful approach to generate new medicines, as demonstrated by the recent clinical approvals of vemurafenib, venetoclax and ribociclib, along with many candidates currently under clinical evaluation.1,2 Despite this promise, progress in FBDD is slowed by problems associated with the overrepresentation of “flat” aromatic compounds and dearth of sp3-rich fragments containing chiral centres in commercial screening libraries.3 Advancement of fragment hits along the pipeline is further hampered by the challenges of synthetically elaborating hit fragments (through growing, linking or merging), often due to the absence of readily available “exit vectors” on fragments of interest, or inability to incorporate new three-dimensional (3D) elements such as stereocentres in a straightforward manner.4,5 Thus, the development of novel methodologies to construct saturated and stereochemically enriched fragment scaffolds in new areas of chemical space is of utmost importance to alleviate these hurdles.6 In particular, it has been noted that strategic application of direct C–H functionalisation chemistries has the potential to significantly streamline these issues.7
Aryl-substituted cyclobutanes and small, fused cyclobutane–heterobicyclic systems constitute prevalent structural entities within pharmaceuticals and natural products (Fig. 1A).8–10 Undeniably, this moiety therefore presents an attractive core ring for a fragment-based library, further evidenced by its small size (four heavy atoms) and potential to act as a constrained heterocycle mimic from a medicinal chemistry perspective.11
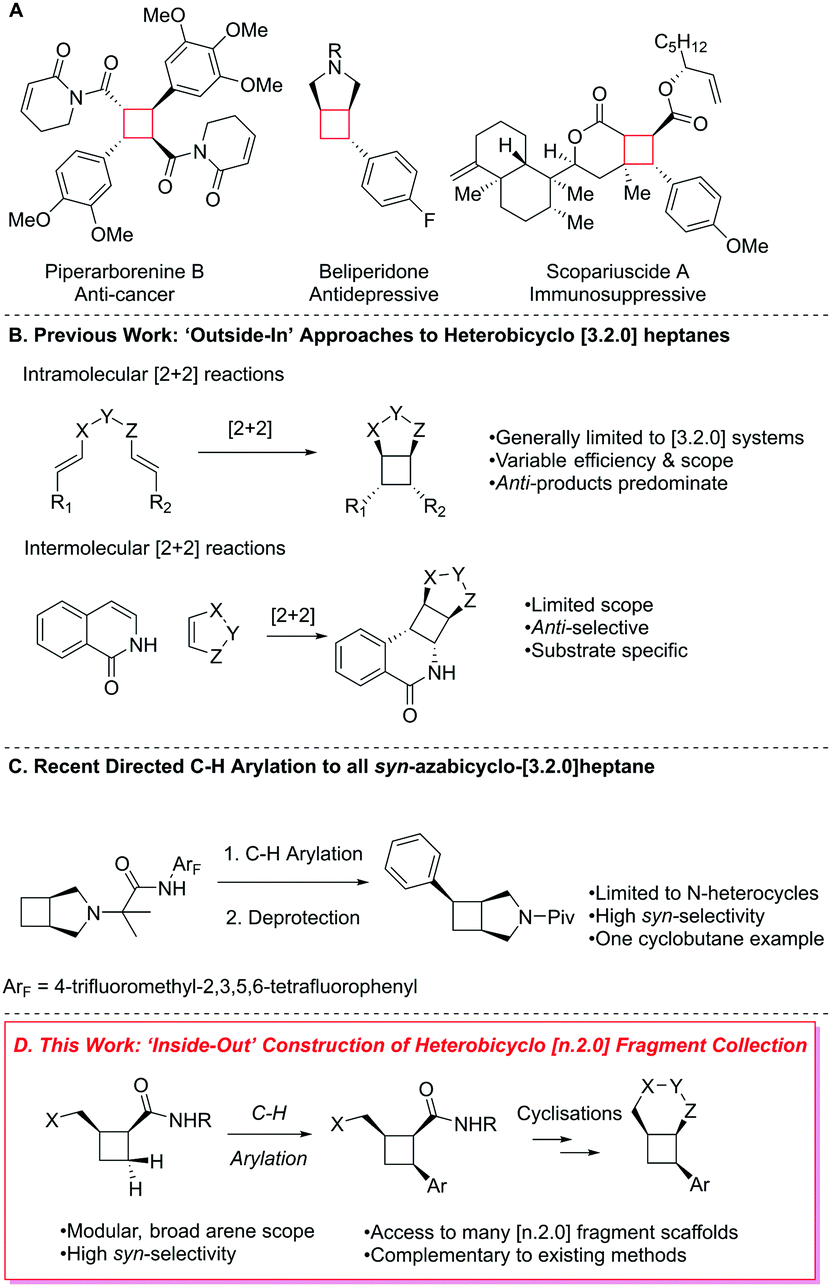 | ||
| Fig. 1 (A) Cyclobutane-containing natural products and pharmaceuticals. (B) Previous work on ‘outside-in’ construction of heterobicyclic systems. (C) Recent application of Pd-catalysed C–H arylation for construction of an all-syn azabicyclo[3 2.0]heptane scaffold. (D) This work, ‘inside-out’ approach to several all-syn heterobicyclic fragments. | ||
Consequently, the construction of cyclobutane-containing bicyclic structures has been the subject of longstanding interest. Commonly, the bicycle is built by constructing the cyclobutane ring via inter- or intramolecular [2+2] reactions, which we term the ‘outside-in’ strategy (Fig. 1B). Intramolecular photochemical [2+2] reactions of tethered 1,6-diene substrates have been frequently employed12 for this purpose. These strategies, however, remain generally limited to the formation of (hetero)bicyclo[3.2.0]heptane scaffolds, proceed with variable levels of efficiency and provide anti-diastereoselectivity (anti-relationship between the fused heterocycle and substituents on the cyclobutane) that can be heavily dependent on the specific substrates employed. Intra- and intermolecular cyclobutane syntheses proceeding via photoredox-type catalytic cycles have also afforded access to certain fused cyclobutane substitution patterns,13 but are often limited in scope and proceed with modest to high anti-diastereoselectivity.
Despite this substantial precedent, a general strategy for the construction of all-syn, fused cyclobutane–heterobicyclo[n.2.0] systems remains elusive. A solution to this problem would represent a significant synthetic advance, particularly in the realm of FBDD, which calls for synthetic solutions to fragment synthesis ideally in numerous 3D arrangements.
The directed, Pd-catalysed C–H arylation reaction first reported by Daugulis in 200514 has now been established as a reliable approach for the stereocontrolled construction of 1,2-syn vicinal stereocentres. This methodology has been extensively utilised for the stereocontrolled C–H arylation of medicinally relevant heterocycles, such as azetidines,15a and pyrrolidinones.15b–d Functionalisation of cyclobutanes under this manifold has been reported in several natural product total syntheses beginning with Baran's synthesis of the piperarborenines in 2011,16 psiguadial B17 as well as scopiariusicide.18 Outside of total synthesis applications, studies focused on the synthesis of functionalised cyclobutanes using this methodology been reported, however, the elaboration into more complex fragments remains limited. Work by the Yu group has detailed the selective formation of all-syn cyclobutyl carboxylic acids and ketones using both conventional and transient directing groups.19 Recently, Sanford and co-workers reported an elegant approach to syn-arylated alicyclic amines via directed, Pd-catalysed transannular C–H arylation (Fig. 1C),20 including a single example of highly syn-selective C–H arylation of the cyclobutane ring in an azabicyclo[3.2.0]heptane system.
Inspired by these pioneering efforts, we envisioned the general construction of all-syn heterobicyclo[n.2.0] fragment systems could be accomplished by exploiting this chemistry using an approach we term the ‘inside-out’ strategy (Fig. 1D). We proposed directed Pd-catalysed C–H arylation on a cyclobutane core, followed by heterocycle formation utilising the latent carboxylic acid oxidation state (accessed via directing group removal) and a proximal heteroatom-containing substituent. Herein, we report the successful realization of this vision towards a novel, sp3-enriched collection of all-syn heterobicyclic fragments in a manner that is stereochemically complementary to most ‘outside-in’ approaches.
Our studies commenced with the construction of a suitable cyclobutane-containing substrate to facilitate exploration of the C–H functionalisation reaction (Fig. 2). Several directing groups have been employed for this purpose, including 2-(methylthio)-aniline (2-MTA),16,21,22 and 8-aminoquinoline,17,23 among others.24,25 In particular, the 2-MTA directing group was selected for this study due to its reported ease of removal, an important factor to enable the downstream derivatisations envisioned. Furthermore, despite its apparent potential during use in the piperarborenine B and scopiariusicide A syntheses, the aryl-iodide scope under a 2-MTA directing group had yet to be systematically explored for cyclobutane C–H arylation on other substrates. Thus, arylation precursor 3 was synthesized in four steps from a commercially available anhydride. Next, we were gratified to find that subjection of 3 and 4-iodoanisole (2 equiv.) to C–H arylation conditions reported by Baran and coworkers16 resulted in a good yield of arylated product 4 with excellent diastereoselectivity.
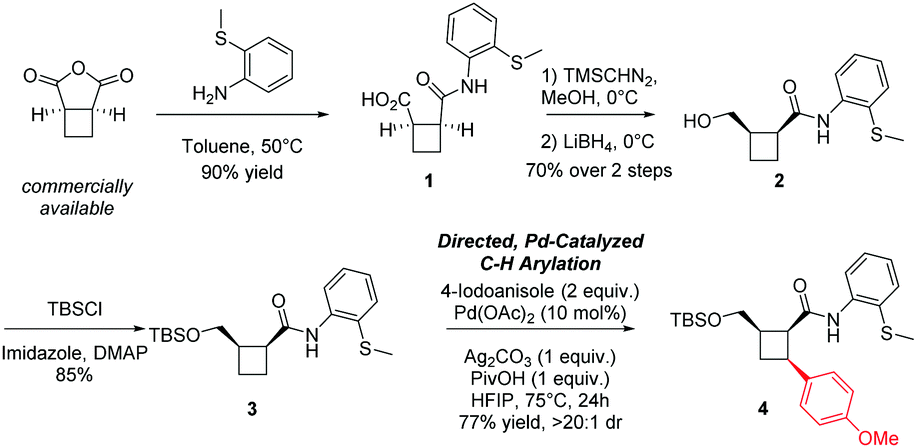 | ||
| Fig. 2 Synthetic sequence to access the all-syn cyclobutane core substrate and initial C–H arylation result. | ||
Finding the C–H arylation conditions fit for purpose, exploration of the aryl-iodide scope began (Fig. 3). The reaction revealed a broad tolerance of electron-rich (4, 5) and electron-poor functional groups (6–15) at both the para- and meta-positions of the arene. Although the literature suggests that ortho-substitution typically is not tolerated in this type of chemistry,23 the use of 2-nitro-4-methoxy-iodobenzene furnished product 16 in 50% yield, providing a useful amine precursor. Incorporation of some pharmaceutically important heterocycles could also be accomplished (17, 18). Interestingly, when 1,4-diiodobenzene was employed as the arene, bis-cyclobutanated product 19 was isolated as the major product. Collectively, these examples demonstrate the utility of the cyclobutane C–H bond as a vector for fragment synthesis. Moreover, the broad range of functional groups incorporated could serve as polar or lipophilic pharmacophores, or as vectors for fragment growth or modification.
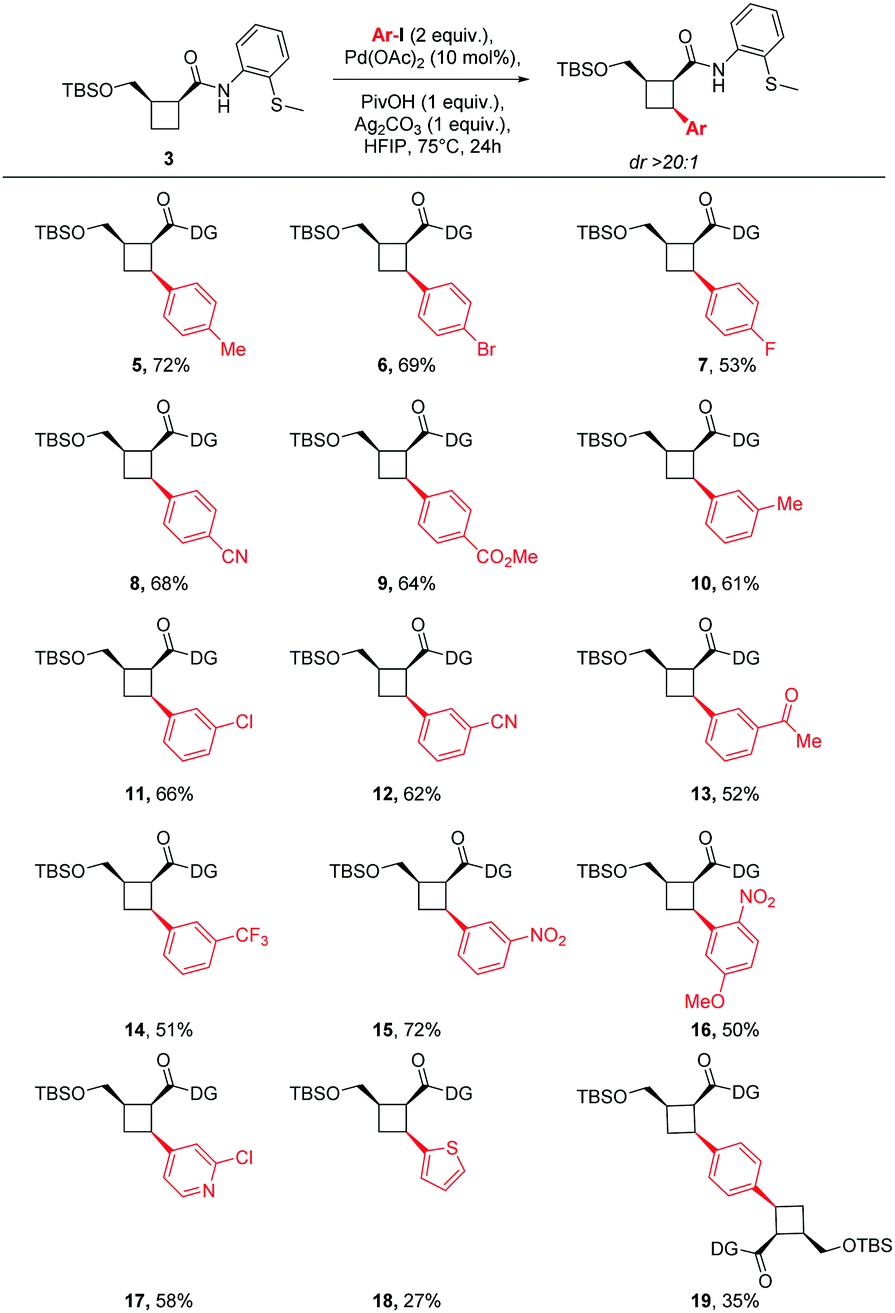 | ||
| Fig. 3 Scope of Pd-catalysed C–H arylation reaction. Reagents and conditions: 3 (1 equiv.), Ar–I (2 equiv.), Pd(OAc)2 (0.1 equiv.), PivOH (1 equiv.), AgzCO3 (1 equiv.), HFIP (0.2 M), 75 °C, 24 h. DG = 2-(methylthio)-aniline. | ||
With these key precursors in hand, we next sought to explore our proposed approach to all-syn heterobicyclo[n.2.0] systems (Fig. 4). To begin, fused butyrolactone–cyclobutane fragment 20 was produced via Boc protection of 4 followed by desilylation with TBAF, resulting in direct lactonisation and displacement of the directing group. This cascade represents an attractive and mild method for directing group removal, which often requires harsh conditions that could epimerise a sensitive stereotriad. This sequence could be readily applied to other arylated products to afford butyrolactones 21–23 (see ESI† for details). Gratifyingly, a single crystal X-ray structure of lactone 20 confirmed the all-syn configuration. Satisfied by this success, the formation of other heterobicyclo[n.2.0] frameworks from butyrolactone 20, were then explored. Pleasinly, reduction of lactone 20 proceeded in straightforward manner to afford diol 24. Subsequent treatment of 24 with triphosgene and pyridine afforded the [5.2.0] 1,3-dioxepane-2-one scaffold 25. Alternatively, reacting 24 with Tf2O and pyridine furnished tetrahydrofuran-containing [3.2.0] scaffold 26. Next, the pyrrolidine-containing [3.2.0] scaffold 27 was constructed by conversion of 24 to a ditosylate followed by heating with benzylamine. Finally, direct opening of lactone 20 with hydrazine followed by Curtius rearrangement afforded cyclic urethane-containing [4.2.0] system 28.
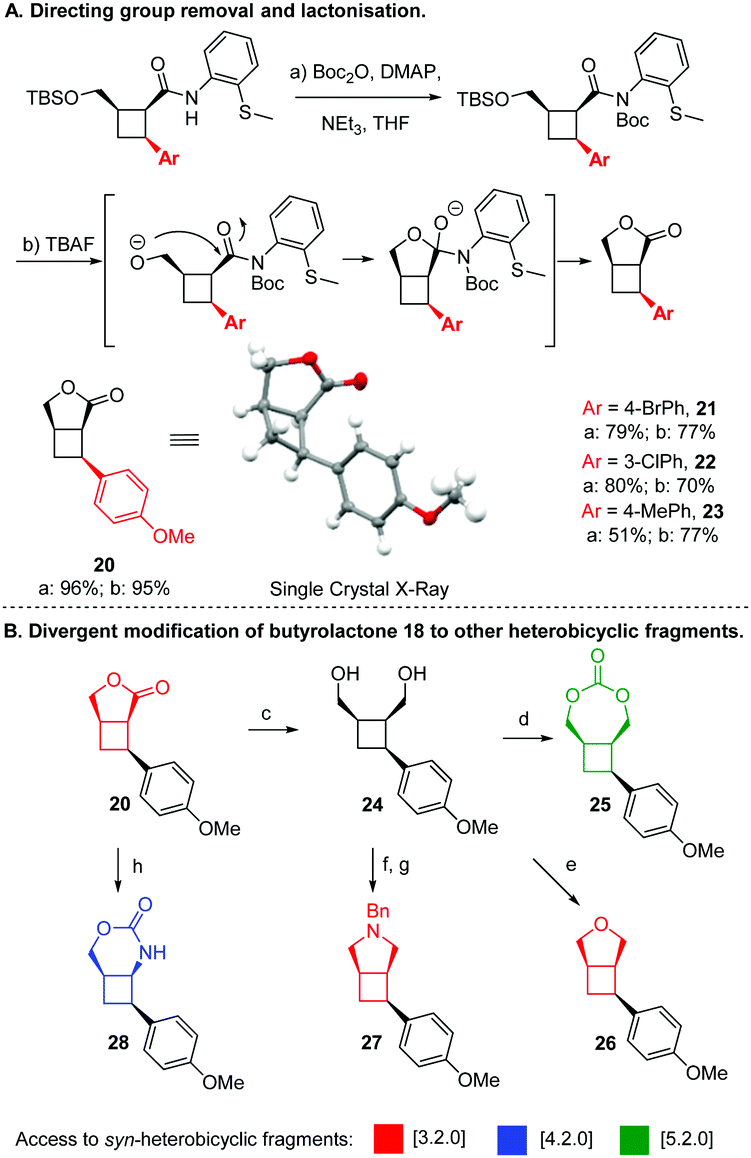 | ||
| Fig. 4 Arylation products were modified in a divergent manner to generate five fused-heterobicyclic scaffolds in a variety of ring sizes. Reagents and conditions: (a) BocjO, DMAP, NEt3, THF, reflux; (b) TBAF. THF. 0 °C to RT; (c) LiAIH4. THF. 0 °C to RT. 96%; (d) triphosgene, pyridine. THF. 0 °C, 66%; (e) Tf2O, pyridine, RT, 85%; (f) TsCI, pyridine, 53%. (g) BnNH2, reflux, 51%; (h) (i) hydrazine hydrate, MeOH, RT. (ii) NaNO2. HCI, THF/H2O, 0 °C. 51% over 2 steps. | ||
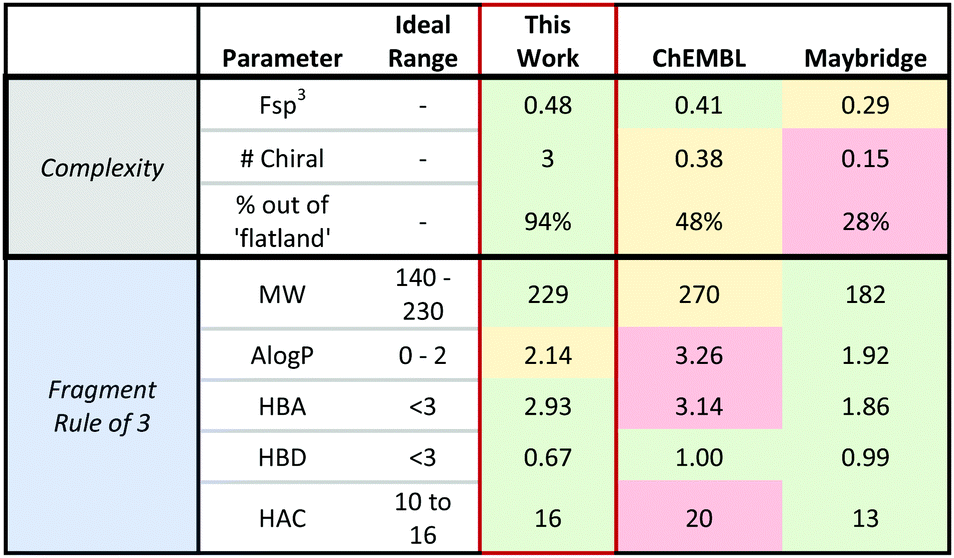
To explore the potential properties of this collection, we created a virtual library of 90 scaffolds using our demonstrated aryl scope (4–19) on each of the final five heterobicyclic scaffolds (20, 25–28) and the diol (24, see ESI† for enumerated products). Cheminformatic analysis was performed, comparing this library to the commercially available Maybridge Rule-of-three (Ro3) core collection26 and representative scaffolds from the ChemBL database (see ESI† for details).27 The all-syn collection demonstrated Ro3 compliance similar to or more favourable than both libraries. Importantly, the all-syn library also exhibited significantly higher complexity metrics (Fsp3, number of chiral centres, and percent of compounds out of ‘flatland’) than the libraries used for comparison. Collectively, these results show that the small molecules produced using the approach outlined in this work can be maximally complex without sacrificing good fragment-like properties (Table 1).
|
There remains a demand for the development of novel methodologies to construct new biologically relevant scaffolds to seed pharmaceutical research. This study has demonstrated that the strategic application C–H arylation of a cyclobutane-containing moiety can enable the synthesis of a collection of all-syn heterobicyclo[n.2.0] containing fragments. This ‘inside-out’ synthetic approach is complementary to ‘outside-in’ approaches relying on [2+2] cycloaddition chemistry for construction of the cyclobutane core, and furnishes fragment precursors in a stereochemically distinct fashion. Cheminformatic analysis of the products compared to commercially available fragments and structurally similar molecules with known biological activity shows the all-syn products to be substantially more complex while maintaining good fragment-like properties. Overall, this work demonstrates the power of merging synthetic strategy, new synthetic methodology, and fragment design principles to facilitate the construction of novel and complex fragments.
This work was supported by the Engineering and Physical Sciences Research Council (EP/P020291/1), Royal Society (Wolfson Research Merit Award) and AstraZeneca. The Spring lab also acknowledges general lab support from the EPSRC, BBSRC, MRC and Royal Society. We thank Dr A. Bond for acquiring the X-ray crystal structure data for compound 20.
X-ray crystallographic data for 20 has been deposited at the CCDC (deposition number 1986749).†
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. W. Murray, D. R. Newell and P. Angibaud, MedChemComm, 2019, 10, 1509–1511 RSC.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- J. A. Johnson, C. A. Nicolaou, S. E. Kirberger, A. K. Pandey, H. Hu and W. C. K. Pomerantz, ACS Med. Chem. Lett., 2019, 10, 1648–1654 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 11, 488–492 CrossRef PubMed.
- S. L. Kidd, T. J. Osberger, N. Mateu, H. F. Sore and D. R. Spring, Front. Chem., 2018, 6, 460 CrossRef PubMed.
- For recent examples in this field see: (a) Y. Wang, J.-Y. Wach, P. Sheehan, C. Zhong, C. Zhan, R. Harris, S. C. Almo, J. Bishop, S. J. Haggarty, A. Ramek, K. N. Berry, C. O’Herin, A. N. Koehler, A. W. Hung and D. W. Young, ACS Med. Chem. Lett., 2016, 7, 852–856 CrossRef CAS PubMed; (b) A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799–6804 CrossRef CAS PubMed; (c) E. Lenci, R. Innocenti, G. Menchi and A. Trabocchi, Front. Chem., 2018, 6, 522 CrossRef CAS PubMed; (d) E. Lenci, A. Rossi, G. Menchi and A. Trabocchi, Org. Biomol. Chem., 2017, 15, 9710–9717 RSC; (e) E. Lenci, G. Menchi, A. Guarna and A. Trabocchi, J. Org. Chem., 2015, 80, 2182–2191 CrossRef CAS PubMed; (f) R. A. Croft, M. A. J. Dubois, A. J. Boddy, C. Denis, A. Lazaridou, A. S. Voisin Chiret, R. Bureau, C. Choi, J. J. Mousseau and J. A. Bull, Eur. J. Org. Chem., 2019, 5385–5395 CrossRef CAS; (g) D. Foley, R. Doveston, I. Churcher, A. Nelson and S. P. Marsden, Chem. Commun., 2015, 51, 11174–11177 RSC; (h) H. Hassan, S. P. Marsden and A. Nelson, Bioorg. Med. Chem., 2018, 26, 3030–3033 CrossRef CAS PubMed; (i) J. Mayol-Llinàs, W. Farnaby and A. Nelson, Chem. Commun., 2017, 53, 12345–12348 RSC; (j) A. R. Hanby, N. S. Troelsen, T. J. Osberger, S. L. Kidd, K. T. Mortensen and D. R. Spring, Chem. Commun., 2020, 56, 2280–2283 RSC.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- I.-L. Tsai, F.-P. Lee, C.-C. Wu, C.-Y. Duh, T. Ishikawa, J.-J. Chen, Y.-C. Chen, H. Seki and I.-S. Chen, Planta Med., 2005, 71, 535–542 CrossRef CAS PubMed.
- Y. Liu, T. Yang, H. Li, M. H. Li, J. Liu, Y. T. Wang, S. X. Yang, J. Zheng, X. Y. Luo, Y. Lai, P. Yang, L. M. Li and Q. Zou, Br. J. Pharmacol., 2013, 168, 632–643 CrossRef CAS PubMed.
- P. R. Verhoest, C. Proulx-Lafrance, M. Corman, L. Chenard, C. J. Helal, X. Hou, R. Kleiman, S. Liu, E. Marr, F. S. Menniti, C. J. Schmidt, M. Vanase-Frawley, A. W. Schmidt, R. D. Williams, F. R. Nelson, K. R. Fonseca and S. Liras, J. Med. Chem., 2011, 54, 7602–7620 CrossRef PubMed.
- (a) S. Poplata, A. Tröster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116(17), 9748–9815 CrossRef CAS PubMed; (b) G. Steiner, R. Munschaner, G. Klebe and L. Siggel, Heterocycles, 1995, 40, 319–330 CrossRef CAS; (c) A. V. Denisenko, T. Druzhenko, Y. Skalenko, M. Samoilenko, O. O. Grygorenko, S. Zozulya and P. K. Mykhailiuk, J. Org. Chem., 2017, 82, 9627–9636 CrossRef CAS PubMed; (d) T. Bach, C. Krüger and K. Harms, Synthesis, 2000, 305–320 CrossRef CAS; (e) D. A. Fort, T. J. Woltering, A. M. Alker and T. Bach, J. Org. Chem., 2014, 79, 7152–7161 CrossRef CAS PubMed; (f) S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948–14951 CrossRef CAS PubMed; (g) S. C. Coote, A. Pöthig and T. Bach, Chem. – Eur. J., 2015, 21, 6906–6912 CrossRef CAS PubMed.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130(39), 12886–12887 CrossRef CAS PubMed; (b) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132(25), 8572–8574 CrossRef CAS PubMed; (c) L. Ackermann, A. Althammer and S. Fenner, Angew. Chem., Int. Ed., 2009, 48, 201–204 CrossRef CAS PubMed; (d) A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991–8994 CrossRef CAS PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- (a) M. Maetani, J. Zoller, B. Melillo, O. Verho, N. Kato, J. Pu, E. Comer and S. L. Schreiber, J. Am. Chem. Soc., 2017, 139, 11300–11306 CrossRef CAS PubMed; (b) D. Antermite, D. P. Affron and J. A. Bull, Org. Lett., 2018, 20, 3948–3952 CrossRef CAS PubMed; (c) D. P. Affron and J. A. Bull, Eur. J. Org. Chem., 2016, 139–149 CrossRef CAS PubMed; (d) D. Antermite and J. A. Bull, Synthesis, 2019, 3171–3204 CAS.
- W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed.
- L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed.
- M. Zhou, X.-R. Li, J.-W. Tang, Y. Liu, X.-N. Li, B. Wu, H.-B. Qin, X. Du, L.-M. Li, W.-G. Wang, J.-X. Pu and H.-D. Sun, Org. Lett., 2015, 17, 6062–6065 CrossRef CAS PubMed.
- (a) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136(22), 8138–8142 CrossRef CAS PubMed; (b) L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 2–9 CrossRef.
- J. Topczewski, P. Cabrera, N. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS PubMed.
- R. A. Panish, S. R. Chintala and J. M. Fox, Angew. Chem., Int. Ed., 2016, 55, 4983–4987 CrossRef CAS PubMed.
- J. L. Hu, L. W. Feng, L. Wang, Z. Xie, Y. Tang and X. Li, J. Am. Chem. Soc., 2016, 138, 13151–13154 CrossRef CAS PubMed.
- R. Parella, B. Gopalakrishnan and S. A. Babu, J. Org. Chem., 2013, 78, 11911–11934 CrossRef CAS PubMed.
- M. Virelli, W. Wang, R. Kuniyil, J. Wu, G. Zanoni, A. Fernandez, J. Scott, M. Vendrell and L. Ackermann, Chem. – Eur. J., 2019, 25, 12712–12718 CrossRef CAS PubMed.
- W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS PubMed.
- N. Mateu, S. L. Kidd, L. Kalash, H. F. Sore, A. Madin, A. Bender and D. R. Spring, Chem. – Eur. J., 2018, 24, 13681–13687 CrossRef CAS PubMed.
- I. Colomer, C. J. Empson, P. Craven, Z. Owen, R. G. Doveston, I. Churcher, S. P. Marsden and A. Nelson, Chem. Commun., 2016, 52, 7209–7212 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1986749. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03237a |
| ‡ Equal contributors. |
| This journal is © The Royal Society of Chemistry 2020 |