
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalysed 3,5-bis-borylation of phthalonitrile enables access to a family of C4h octaarylphthalocyanines†
Katie D.
Mulholland
a,
Sangbin
Yoon
 a,
Christopher C.
Rennie
b,
Eleanor K.
Sitch
a,
Alasdair I.
McKay
c,
Katharina
Edkins
d and
Robert M.
Edkins
*b
a,
Christopher C.
Rennie
b,
Eleanor K.
Sitch
a,
Alasdair I.
McKay
c,
Katharina
Edkins
d and
Robert M.
Edkins
*b
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
bWestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: robert.edkins@strath.ac.uk
cSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
dDivision of Pharmacy and Optometry, University of Manchester, Stopford Building, Manchester M13 9PT, UK
First published on 18th June 2020
Abstract
Ir-catalysed borylation of phthalonitrile produces both 4-(Bpin)phthalonitrile (1) and 3,5-bis(Bpin)phthalonitrile (2), which are potential divergent intermediates for the synthesis of functionalized phthalocyanines. To exemplify the utility of 2, we have prepared a series of 3,5-bis-arylphthalonitriles that in turn undergo sterically controlled regioselective cyclotetramization to give previously unknown C4h 1,3,8,10,15,17,22,24-octaarylphthalocyanines.
Phthalocyanines (Pcs) have been extensively explored over recent decades for their use in dye-sensitized solar cells,1–3 single-molecule magnets,4,5 (photo)catalysis,6 and various cancer phototherapies,7–9 making them one of the most important classes of synthetic chromophores.
Pcs are commonly synthesized by the cyclization of four substituted phthalonitrile precursors around a metal-ion template. A recurring issue in Pc chemistry is that the synthesis of substituted phthalonitrile precursors is often lengthy and hindered by the low reactivity of electron-poor phthalonitrile in SEAr reactions. Here, we report our initial investigation into the use of sterically controlled Ir-catalysed C–H borylation10–12 to functionalize phthalonitrile that circumvents its unfavourable electronics. This study was further motivated by the ease with which aryl boronic acid pinacol ester (Bpin) groups introduced by this reaction might, in general, be converted to a wide range of other functional groups, including various amines, ethers, thioethers, (hetero)arenes, or to an azide, halide, nitro or alcohol group for further functionalization using known transformations.13 Such conversions can often be performed using one-pot borylation-functionalization methodologies, such as demonstrated by the cyanation of in situ generated arylboronic esters14 or their conversion to perfluoroalkyl groups,15 which would make this a versatile route for preparing substituted phthalonitriles.
Based on the steric, rather than electronic selectivity of Ir-catalysed borylation, it was expected that borylation of phthalonitrile using 1.0 eq. B2pin2 (pin = pinacolato), 1.5 mol% [Ir(COD)(OMe)]2 and 3.0 mol% dtbpy (dtbpy = 4,4′-bis(tBu)-2,2′-bipyridyl) in methyl tert-butyl ether (MTBE) at room temperature would afford 1,2-dicyano-4-(Bpin)benzene, 1, selectively (Scheme 1). This reaction did indeed proceed with quantitative consumption of phthalonitrile to give 1 as the major product; however, traces of the bis-borylated 3,5-bis(Bpin)-1,2-dicyanobenzene (2) were also observed by 1H NMR spectroscopy. Borylation ortho to the relatively low steric-demand cyano group in the absence of more sterically accessible sites, as observed here in the bis-borylation reaction to give unpredicted compound 2, has been reported for para-substituted benzonitriles16 and was also observed during the borylation of 2-methylbenzonitrile, where 2,4-bis(Bpin)-6-methylbenzonitrile was obtained as a minor by-product.17 We also note that arylnitrile groups are competent directing groups for a range of metal-catalysed reactions.18–21
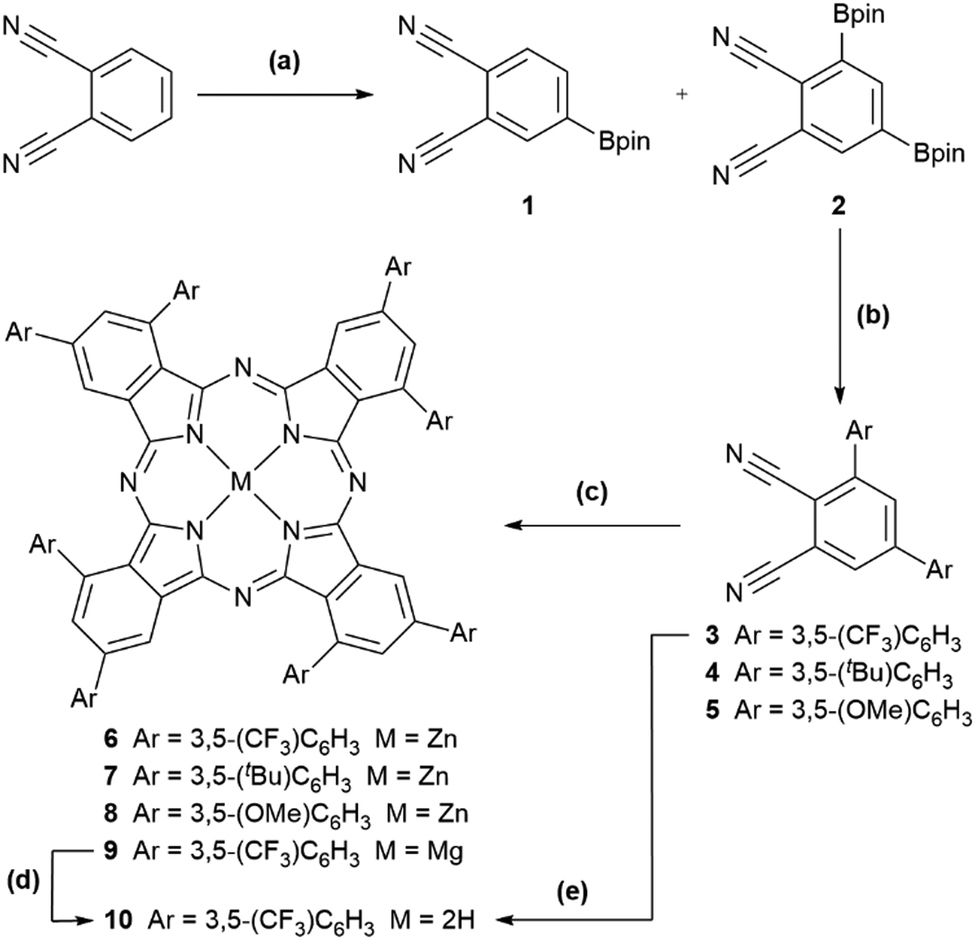 | ||
Scheme 1 Synthesis of 6–10 starting from phthalonitrile. (a) Ir-catalysed borylation: 1.0 eq. B2pin2, 1.5 mol% [Ir(COD)(OMe)]2, 3.0 mol% dtbpy, MTBE, 1.0 M, r.t., 24 h gave 1 with trace 2; same conditions except 1.1 eq. B2pin2, 55 °C gave 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 1:2; same conditions except 1.5 eq. B2pin2, 55 °C, 1.3 M gave 2 only [46%]. (b) Suzuki–Miyaura cross-coupling: 2.4 eq. 3,5-bis(R)bromobenzene (R = CF3, tBu, OMe for 3, 4, and 5), 5 mol% Pd2(dba)3, 20 mol% SPhos, 4.0 eq. CsF, 1,4-dioxane, 65 °C [19% (3), 50% (4), and 21% (5)]. Yield of 3 increased to 68% using 4.0 eq. 3,5-bis(CF3)iodobenzene, 4.0 eq. Cs2CO3. (c) Macrocyclization: 0.2–1.1 eq. Zn(OAc)2·2H2O (6–8) or Mg(OAc)2·4H2O (9), 1-pentanol, 1.0 eq. DBU, 132 °C [39% (6), 25% (7), 13% (8), 5% (9)]. (d) Demetallation: acetic acid, 110 °C, 2 h, quantitative by UV-visible absorption spectroscopy. (e) One-pot macrocyclization/demetallation: (i) as (c) with Mg(OAc)2·4H2O; (ii) HCl (1 M, 5 mL), 60 °C, 16 h [5%]. All yields are isolated. See ESI† for further details. :7 1:2; same conditions except 1.5 eq. B2pin2, 55 °C, 1.3 M gave 2 only [46%]. (b) Suzuki–Miyaura cross-coupling: 2.4 eq. 3,5-bis(R)bromobenzene (R = CF3, tBu, OMe for 3, 4, and 5), 5 mol% Pd2(dba)3, 20 mol% SPhos, 4.0 eq. CsF, 1,4-dioxane, 65 °C [19% (3), 50% (4), and 21% (5)]. Yield of 3 increased to 68% using 4.0 eq. 3,5-bis(CF3)iodobenzene, 4.0 eq. Cs2CO3. (c) Macrocyclization: 0.2–1.1 eq. Zn(OAc)2·2H2O (6–8) or Mg(OAc)2·4H2O (9), 1-pentanol, 1.0 eq. DBU, 132 °C [39% (6), 25% (7), 13% (8), 5% (9)]. (d) Demetallation: acetic acid, 110 °C, 2 h, quantitative by UV-visible absorption spectroscopy. (e) One-pot macrocyclization/demetallation: (i) as (c) with Mg(OAc)2·4H2O; (ii) HCl (1 M, 5 mL), 60 °C, 16 h [5%]. All yields are isolated. See ESI† for further details. | ||
By increasing the reaction temperature to 55 °C and the amount of B2pin2 to 1.1 eq., 70% bis-borylation, 30% mono-borylation and quantitative consumption of phthalonitrile was achieved. Sublimation (150 °C/0.3 mbar) separated 1 from the crude mixture, while recrystallization of the residue from MTBE provided 2. The structures of 1·0.25H2O and 2 were obtained by single-crystal X-ray diffraction (SC-XRD) (Fig. S22, ESI†). Increasing the amount of B2pin2 to 1.5 eq. and using a higher reaction concentration (1.3 M vs. 1.0 M) increased conversion to 2 (46% isolated yield) and removed the need for the sublimation step.
With the unexpected product 2 in hand, we decided to consider first its potential for the preparation of phthalocyanines for the following reasons. A common problem in the synthesis of functionalized Pcs is that reaction of substituted phthalonitriles lower than C2v symmetry usually produces a 1:4:2:1 statistical, and often inseparable, mixture of C4h, Cs, C2v, and D2h regioisomers.22 Regardless of which function or application is sought, formation of a single Pc isomer is desirable due to their potentially differing physical, optical and biological properties. In the rare cases where separation of one or more of these four isomers has been possible, it has been necessary to use bespoke HPLC columns23,24 or repeated column chromatography,25 limiting the generality of the procedure. Recrystallization has occasionally been successful as part of a multi-step purification of the C4h isomer.26,27 If, however, bulky substituents, e.g. substituted phenyl rings,28,29 branched alkoxides,24,26,30 amines,27,31 or trialkylsilyl groups,32 are introduced at the 3-position of the phthalonitrile precursor, then exclusive formation of the C4h Pc isomer can sometimes be enforced through steric control or the number of isomers formed in the mixed A/B phthalonitrile synthesis of A3B-33,34 and ABAB-type35,36 Pcs can be reduced. The substituted phthalonitriles used in these previous studies were prepared by cross-coupling of 3-(OTf)phthalonitrile, nucleophilic aromatic substitution of 3-nitrophthalonitrile, or directed ortho-lithiation of 4-alkylphthalonitriles, respectively. While successful, these previous reports have been limited in allowing the introduction of further functionalization on to the phthalonitrile, either due to the difficulty of synthesizing the substituted phthalonitriles via traditional routes or the incompatibility of desired substituents with organolithium reagents. With this in mind, 2 was seen as a potential precursor to 3,5-bis-substituted phthalonitriles, which could include derivatives having a bulky substituent in the 3-position to direct the regioselective synthesis of C4h phthalocyanines through steric control while also bearing a second substituent in the 5-position.
As a first demonstration of the potential utility of 2 as a divergent intermediate in the synthesis of functionalized regioregular Pcs, a series of 3,5-bis(aryl)phthalonitriles 3–5 with varying steric demand and electronic character was prepared from 2 by Suzuki–Miyaura cross-coupling with 3,5-bis(R)bromobenzenes (R = CF3, tBu, OMe, respectively). 5 mol% Pd2(dba)3 pre-catalyst, 20 mol% SPhos ligand and 4 eq. CsF base in 1,4-dioxane at 65 °C gave 3–5 in moderate yields (19–50%). Using 3,5-bis(CF3)iodobenzene and Cs2CO3 as the base improved the yield of 3 from 19 to 68%. The SC-XRD structures of 3–5 are shown in Fig. S22 (ESI†). These products were expected to undergo regioselective cyclotetramization to the previously unexplored C4h 1,3,8,10,15,17,22,24-octaarylphthalocyanine family of compounds.
Reaction of 3 with Zn(OAc)2·2H2O in 1-pentanol in the presence of DBU indeed afforded 1,3,8,10,15,17,22,24-octaarylphthalocyanine 6. TLC showed only a single dark-green compound, which could be separated by recrystallization in 39% yield, with the mass balance likely being oligomeric by-products. Fig. 1 shows partial 1H and 19F{1H} NMR spectra of 6, which confirm the high symmetry of the Pc by the single set of sharp resonances for the six aryl proton environments and two inequivalent CF3 environments, respectively. Only the C4h and D2h isomers would show this equivalence of the rings; these are the least and most sterically encumbered isomers.
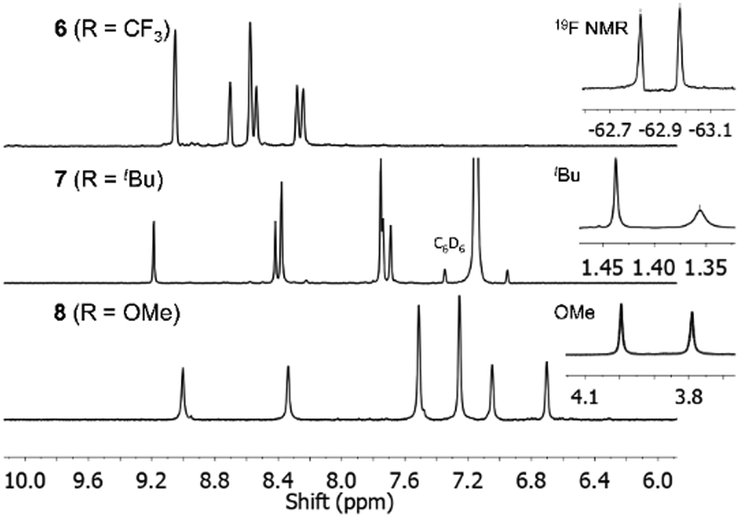 | ||
| Fig. 1 Partial 1H NMR spectra of 6–8 and 19F{1H} NMR spectrum of 6. The spectra are consistent with the high symmetry of each Pc. | ||
Pcs 7 and 8, starting from precursors 4 and 5, were synthesized analogously in isolated yields of 25 and 13%, respectively. Sharp, well-defined 1H NMR spectra were obtained for 7 and 8 with six aromatic proton environments clearly observed (Fig. 1).
The SC-XRD structure of 6, obtained after recrystallization from acetone/pyridine, confirmed the nominal C4h symmetry of 6, although the Zn atom is additionally ligated by a 3:1 mixture of pyridine and water (pyridine adduct 6·NC5H5 shown in Fig. 2). Distortion of the Pc ring away from planarity can be seen, with a maximum fold angle of 6.79(9)° for benzo group 1 relative to the meso-N4 plane (N1, N2 and symmetry equivalents). The coordinated Zn atom is 0.49 Å out of the Pc meso-N4 plane and is disordered 50:50 either side of the ring. The α-substituents, rings B/D, have mean-plane dihedral angles of 50.49(10)° and 56.30(12)° with respect to adjoining benzo groups 1 and 2, respectively. These large twist angles prevent neighbouring α-substituents from overlapping (unlike when unsubstituted α-phenyl groups are used, which can overlap with distortion of the Pc ring37), enforcing the formation of the single C4h isomer. Peripheral β-position rings A/C have dihedral angles of 49.0(1)° and 40.6(1)°.
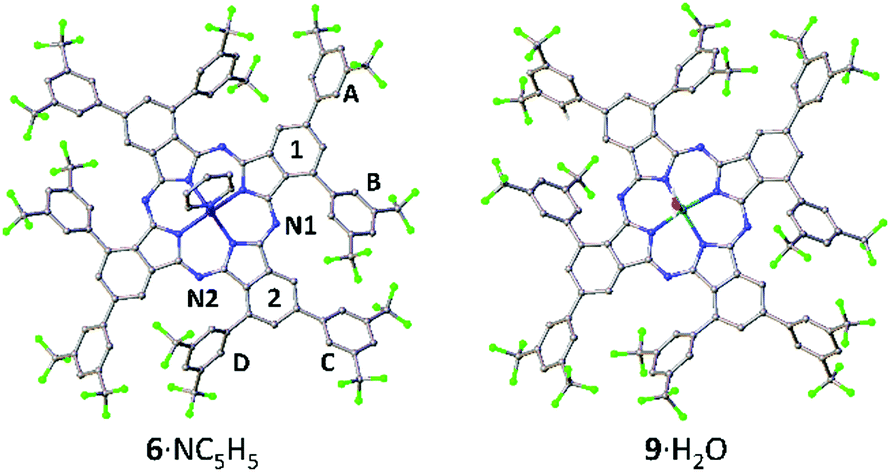 | ||
| Fig. 2 Structures of 6·NC5H5 and 9·H2O from SC-XRD. Disorder and hydrogen atoms are omitted for clarity. Analogous ring labelling to that of 6 is used for all structures. | ||
The SC-XRD structure of 7 was obtained following recrystallization from acetone (Fig. S23, ESI†); however, the quality of this structure is low, due to a large amount of incorporated disordered solvent, the rotational disorder of the tBu groups of the two crystallographically independent Pcs 7(A) and 7(B), and the weak diffraction of the crystal. Nonetheless, the Pc rings and the eight aryl substituents of both 7(A) and 7(B) were unambiguously refined and the structure conclusively confirms the C4h symmetry of both molecules. Ligation of the Zn atoms can only be defined conclusively for 7(B) and has been resolved as a water molecule. The large size of the 3,5-bis(tBu)phenyl groups means that the β-positions are not free to rotate due to close proximity of neighbouring α-substituents, i.e. groups A/D and B/C orient concertedly. In comparison, the smaller 3,5-bis(trifluoromethyl)phenyl groups of 6 show less correlation and thus have less restricted rotation.
C 4h MgPc 9 was synthesized analogously to 6 in an unoptimized 5% yield by reaction of 3 with Mg(OAc)2·4H2O. The SC-XRD structure of 9 (recrystallized from acetone/hexane, Fig. 2) has disordered solvent in the voids between the cup-like Pcs, but the Pc structure itself is well resolved and the symmetry confirmed. The Mg atom has a ligand that was resolved as water, and sits 0.74 Å out of the Pc plane. The α-substituents have large dihedral angles of 54.8(3)° and 49.0(3)° for B and D respectively. The β-aryl rings A and C are twisted by 36.6(3)° and 51.8(3)°. Similar to 6, there is no correlation between dihedral angles of close contacting rings: B/C rings twist in the opposite sense, while A/D differ in dihedral angle by about 12° and are therefore assumed to have some rotational freedom.
Successful de-metalation of 9 to free-base Pc 10 by heating in acetic acid was monitored by UV-visible absorption and 1H/19F NMR spectroscopies. Alternatively, 10 can be synthesized in 5% yield with a one-pot procedure using HCl to demetalate the intermediate MgPc. The split Q band in the UV-visible absorption spectrum of 10 confirmed symmetry reduction from C4h to nominal C2h (Fig. S22, ESI†). As free-base Pcs can be metalated with a range of metals other metals could be introduced into these regioregular 1,3,8,10,15,17,22,24-octaarylphthalocyanines.38
The presence of eight aryl ring substituents on 6–9 makes them highly soluble in organic solvents of different polarity and coordinating ability, e.g. toluene, CH2Cl2, CHCl3, THF, acetone, pyridine, and MeCN. The extinction coefficient of 6 at the Q-band maximum (694 nm) is large (3.8 × 105 M−1 cm−1, acetone solution). Normalized UV-visible absorption spectra of 6 in acetone solution in the measurable concentration range of 1.7 × 10−7 to 1.7 × 10−4 M were identical within experimental error (Fig. S25, ESI†), indicating that 6 does not aggregate at these concentrations, unlike many common Pc derivatives. The introduction of the eight aryl groups around the Pc core in a regioregular fashion is therefore an effective strategy to inhibit interactions through π-stacking, with the large dihedral angles of the α-substituents being especially beneficial. As aggregation of Pcs often leads to quenching of excited states, and thus lower fluorescence and singlet-oxygen quantum yields, minimizing aggregation is beneficial for most applications.39
The Q00 bands in the absorption spectra of 6–8 are single peaks (Fig. 3), rather than split into Qx/Qy components, consistent with their C4h symmetry and the two-fold degeneracy of the LUMO. Near-infrared emitting 6–9 (λmax = 701–717 nm) have small Stokes shifts (260–280 cm−1) and fluorescence lifetimes of ca. 1.9–5.4 ns (Fig. 3, Table 1 and Fig. S24, S26–S31, ESI†). The fluorescence and singlet-oxygen quantum yields of 6 are 0.17 and 0.67, respectively. Thus, encouragingly for potential applications, the eight aryl substituents of 6–9 only lead to a minor increase in non-radiative decay, despite the additional rotational freedom relative to the parent ZnPc, and the triplet state is still sufficiently energetic to sensitize singlet oxygen efficiently. Both the absorption and emission spectra of 7 are significantly broader than 6, 8 or 9; the hindered rotation of the aryl groups of 7 observed in its SC-XRD structure may be causing inhomogeneous broadening, i.e. there are different solution conformations of 7 that interconvert relatively slowly due to steric crowding. This suggests that having 3,5-bis(tBu)aryl groups in both the α and β-positions is close to the steric crowding limit for successful synthesis of 1,3,8,10,15,17,22,24-octaarylphthalocyanines.
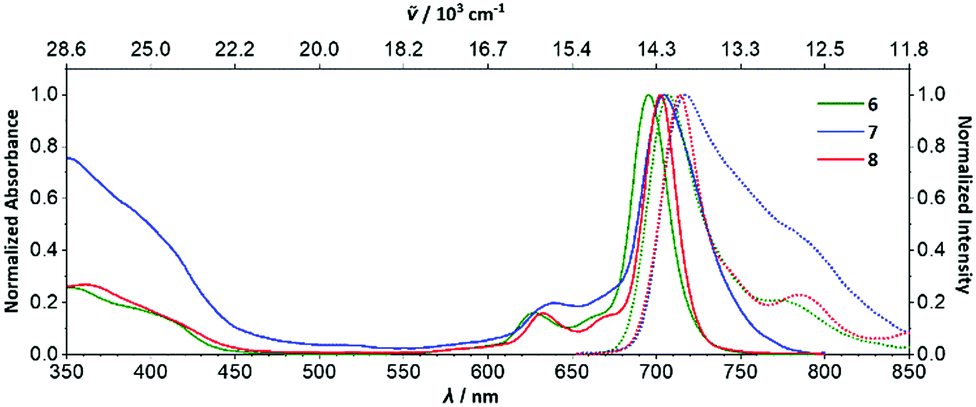 | ||
| Fig. 3 Normalized absorption (solid lines) and emission (dotted lines) spectra of 6–8 in acetone solution. | ||
In conclusion, we report 4-(Bpin)- and 3,5-bis(Bpin)phthalonitrile (1 and 2, respectively), synthesized by Ir-catalysed C–H mono- and unpredicted bis-borylation of phthalonitrile, as potential divergent intermediates for phthalocyanine chemistry. As a first demonstration of the utility of 2, we synthesized a series of 3,5-substituted phthalonitrile derivatives bearing bulky aryl groups that subsequently undergo regioselective cyclization to afford a series C4h 1,3,8,10,15,17,22,24-octaarylphthalocyanines, 6–10, as confirmed by NMR spectroscopy and by SC-XRD for 6, 7 and 9. The high symmetry of these non-aggregating Pc derivatives was further confirmed by UV-visible absorption spectroscopy. We are currently investigating the use of 1 in preparing phthalonitrile derivatives, as well as exploring methods to differentiate the two Bpin groups of 2 to facilitate the synthesis of multifunctional phthalocyanines with controlled symmetry. The rapid functionalization of phthalonitrile reported herein may also find use in the preparation of near-infrared azaBODIPY dyes, potentially further extending its usefulness.42,43
R. M. E. thanks the Royal Commission for the Exhibition of 1851 for a research fellowship, the John Fell Fund of the University of Oxford for a grant, the Analytical Chemistry Trust Fund for a summer studentship to S. B. Y., and AllyChem Co., Ltd for a gift of B2pin2. R. M. E. is grateful to Prof. Stephen Faulkner (University of Oxford) for providing laboratory space and generous support during his 1851 Fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- P. Brogdon, H. Cheema and J. H. Delcamp, ChemSusChem, 2018, 11, 86–103 CrossRef CAS PubMed.
- M. Urbani, M.-E. Ragoussi, M. K. Nazeeruddin and T. Torres, Coord. Chem. Rev., 2019, 381, 1–64 CrossRef CAS.
- K. Katoh, H. Isshiki, T. Komeda and M. Yamashita, Coord. Chem. Rev., 2011, 255, 2124–2148 CrossRef CAS.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685 CrossRef CAS PubMed.
- X. Li, X.-H. Peng, B.-D. Zheng, J. Tang, Y. Zhao, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, Chem. Sci., 2018, 9, 2098–2104 RSC.
- X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- J. S. Wright, P. J. H. Scott and P. G. Steel, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202001520.
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, 2nd edn, 2011 Search PubMed.
- C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389–11391 CrossRef CAS PubMed.
- N. D. Litvinas, P. S. Fier and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 536–539 CrossRef CAS PubMed.
- G. A. Chotana, M. A. Rak and M. R. Smith, J. Am. Chem. Soc., 2005, 127, 10539–10544 CrossRef CAS PubMed.
- H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505–3515 RSC.
- W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362–8366 CrossRef CAS PubMed.
- B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786–2791 CrossRef CAS PubMed.
- M. C. Reddy and M. Jeganmohan, Chem. Commun., 2015, 51, 10738–10741 RSC.
- Y. Ping, L. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2017, 359, 3274–3291 CrossRef CAS.
- For simplicity, we approximate here and throughout that the orientations of the aryl substituents and any Pc ring distortion do not lower the symmetry and thus the point groups are determined only by the positions of the substituents around the Pc ring.
- M. Sommerauer, C. Rager and M. Hanack, J. Am. Chem. Soc., 1996, 118, 10085–10093 CrossRef CAS.
- M. Durmuş, S. Yeşilot and V. Ahsen, New J. Chem., 2006, 30, 675–678 RSC.
- V. Novakova, J. Roh, P. Gela, J. Kuneš and P. Zimcik, Chem. Commun., 2012, 48, 4326–4328 RSC.
- W. Liu, C.-H. Lee, H.-W. Li, C.-K. Lam, J. Wang, T. C. W. Mak and D. K. P. Ng, Chem. Commun., 2002, 628–629 RSC.
- Y. Chen, W. Fang, K. Wang, W. Liu and J. Jiang, Inorg. Chem., 2016, 55, 9289–9296 CrossRef CAS PubMed.
- J. Ranta, T. Kumpulainen, H. Lemmetyinen and A. Efimov, J. Org. Chem., 2010, 75, 5178–5194 CrossRef CAS PubMed.
- N. Iida, E. Tokunaga, N. Saito and N. Shibata, J. Fluorine Chem., 2014, 168, 93–98 CrossRef CAS.
- M. Canlıca, J. Mol. Struct., 2020, 1214, 128160 CrossRef.
- S. Yamamoto, K. Kuribayashi, T. N. Murakami, E. Kwon, M. J. Stillman, N. Kobayashi, H. Segawa and M. Kimura, Chem. – Eur. J., 2017, 23, 15446–15454 CrossRef CAS PubMed.
- N. Iida, K. Tanaka, E. Tokunaga, H. Takahashi and N. Shibata, ChemistryOpen, 2015, 4, 102–106 CrossRef CAS PubMed.
- L. Tejerina, M. V. Martínez-Díaz and T. Torres, Org. Lett., 2015, 17, 552–555 CrossRef CAS PubMed.
- L. Tejerina, M. V. Martínez-Díaz, M. K. Nazeeruddin and T. Torres, Chem. – Eur. J., 2016, 22, 4369–4373 CrossRef CAS PubMed.
- E. Fazio, J. Jaramillo-García, G. de la Torre and T. Torres, Org. Lett., 2014, 16, 4706–4709 CrossRef CAS PubMed.
- E. Fazio, J. Jaramillo-García, M. Medel, M. Urbani, M. Grätzel, M. K. Nazeerudin, G. de la Torre and T. Torres, ChemistryOpen, 2017, 6, 121–127 CrossRef CAS PubMed.
- T. Fukuda, K. Ono, S. Homma and N. Kobayashi, Chem. Lett., 2003, 32, 736–737 CrossRef CAS.
- C. C. Leznoff, S. M. Marcuccio, S. Greenberg, A. B. P. Lever and K. B. Tomer, Can. J. Chem., 1985, 63, 623–631 CrossRef CAS.
- N. Kobayashi and A. B. P. Lever, J. Am. Chem. Soc., 1987, 109, 7433–7441 CrossRef CAS.
- L. De Boni, E. Piovesan, L. Gaffo and C. R. Mendonça, J. Phys. Chem. A, 2008, 112, 6803–6807 CrossRef CAS PubMed.
- A. C. Beveridge, B. A. Bench, S. M. Gorun and G. J. Diebold, J. Phys. Chem. A, 2003, 107, 5138–5143 CrossRef CAS.
- R. Gresser, M. Hummert, H. Hartmann, K. Leo and M. Riede, Chem. – Eur. J., 2011, 17, 2939–2947 CrossRef CAS PubMed.
- V. Bandi, H. B. Gobeze and F. D'Souza, Chem. – Eur. J., 2015, 21, 11483–11494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols and other experimental details, copies of NMR spectra, crystallographic data, and absorption and emission spectra. CCDC 1989433–1989438, 1989726 and 1989727. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03161e |
| This journal is © The Royal Society of Chemistry 2020 |