
Efficient formation of [3]pseudorotaxane based on cooperative complexation of dibenzo-24-crown-8 with diphenylviologen axle†
Yoko
Sakata
 *ab,
Takaya
Ogura
a and
Shigehisa
Akine
*ab
*ab,
Takaya
Ogura
a and
Shigehisa
Akine
*ab
aGraduate School of Natural Science and Technology, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan. E-mail: sakata@se.kanazawa-u.ac.jp; akine@se.kanazawa-u.ac.jp
bWPI Nano Life Science Institute (WPI-NanoLSI), Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan
First published on 9th June 2020
Abstract
A novel cooperative [3]pseudorotaxane system consisting of dibenzo-24-crown-8 (DB24C8) and diphenylviologen axle has been developed. The two-step formation of the [3]pseudorotaxane occurred in a positive-cooperative manner. The corresponding [3]rotaxane was successfully obtained from just a stoichiometric mixture of each component by end-capping without dissociation.
Among the various kinds of mechanically-interlocked molecules,1 rotaxane derivatives have highly contributed to the development of new types of molecular machines, such as molecular shuttles,2 a molecular elevator,3 and a molecular muscle.4 The dynamics of the [2]rotaxanes originates from rotating and shuttling motions e.g., the change in the relative positions of the wheel and axle. Oligorotaxanes, such as [3]rotaxanes, which have an increased number of wheel components, can be good candidates to enable more complex motions, because they are expected to undergo a variety of movements such as the changes in the relative positions or orientations of the two or more wheel components.5
The general strategy to obtain [3]rotaxanes requires an axle molecule that has two station units so that each station unit can bind to one wheel component. In fact, [3]rotaxanes are easily available when the wheel-station interactions are strong enough, as seen in those based on metal-coordination.6 If the interaction is not strong enough, however, the formation of [3]rotaxane becomes less efficient. The synthetic attempts often produce a mixture of [2]- and [3]rotaxanes unless a large excess amount of the wheel is used.7 This is because the second wheel threading is less favourable than the first one due to a steric/electrostatic repulsion. This is interpreted as a negative-cooperative process, but if the two-step complexation occurs in a positive-cooperative fashion,8 [3]rotaxanes would be more efficiently synthesized, thus allowing us to use them as a convenient motif for dynamic functional molecules. Actually, positive cooperativity was observed for the formation of several [3]pseudorotaxanes or [3]rotaxanes containing a cryptand,9 shape-persistent macrocycles, etc.10 as the wheel units. Since crown ethers have been key players in rotaxane chemistry,1–3 it is desirable to develop cooperative [3]pseudorotaxane systems based on crown ethers, which could be a versatile intermediate for the synthesis of [3]rotaxanes.
Recently, the crystal structure of a [3]pseudorotaxane, MV·(DB24C8)2, in which two crown ether wheels simultaneously interact with one dimethylviologen (MV) molecule, has been reported.11 This was somewhat surprising because, previously, the MV motif had solely been used to obtain [2]pseudorotaxane structures, that is, only one wheel molecule interacts with the MV station of the rotaxane axle through π–π and/or CT interactions between the electron-rich alkoxybenzene unit and the electron-deficient bipyridinium unit.2,3,12 In this unique [3]pseudorotaxane MV·(DB24C8)2, however, the binding with the second wheel was reported to be weak so that only [2]pseudorotaxane was observable in solution. If the formation efficiency is improved, this [3]pseudorotaxane motif could be much more attractive not only for its unique structural feature but also for the redox behaviour of the viologen axles. In this study, we explored the [3]pseudorotaxane formation behaviour of the two MV analogues, A1 and A2, having phenyl and benzyl groups, respectively, which could enhance the MV-crown ether interactions in solution (Fig. 1). We discovered that the combination of diphenylviologen A1 and DB24C8 exhibits an unexpectedly highly positive cooperativity during the [3]pseudorotaxane formation. We successfully applied this positive-cooperative binding to the efficient synthesis of [3]rotaxane by end-capping of the carboxy groups at the two terminals of the axle molecule.
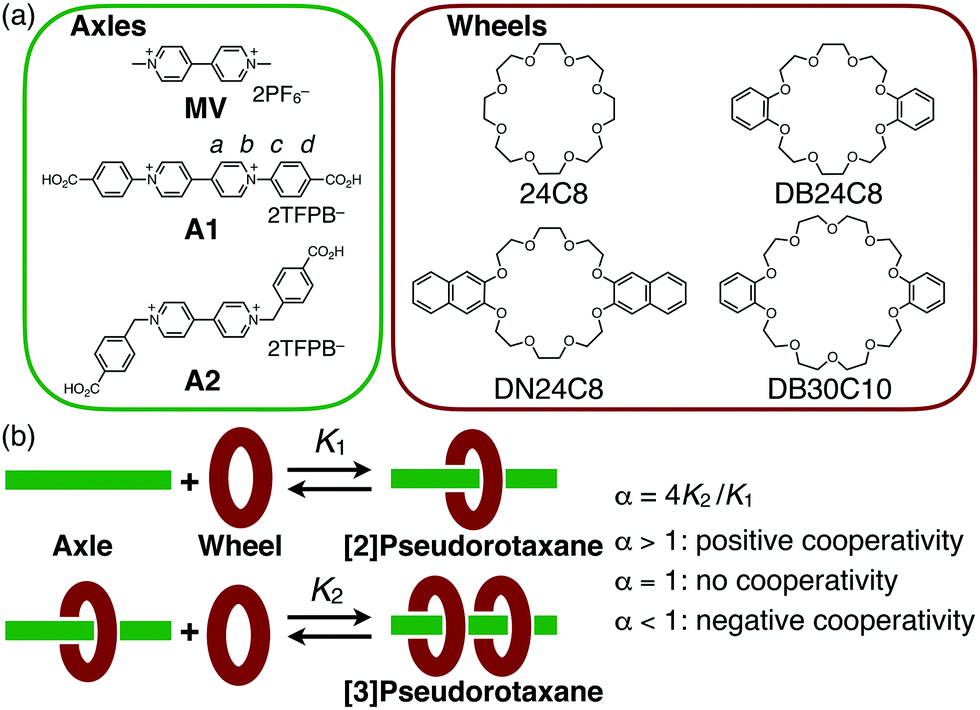 | ||
| Fig. 1 (a) Chemical structures of axle and wheel molecules used in this study (TFPB− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate). (b) Equilibria for the formation of [3]pseudorotaxane and definition of the binding constants K1, K2 and the cooperativity factor α. | ||
We used two kinds of viologen axles, diphenylviologen A1 and dibenzylviologen A2 as TFPB salts (TFPB− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate),13 which were prepared from the corresponding halide salts14,15 (Fig. 1a). Two carboxy groups are introduced at the two terminals as reaction sites for conversion to the corresponding [3]rotaxane by end-capping. As the wheel units, four kinds of crown ether derivatives, 24C8, DB24C8, DN24C8, and DB30C10, were used.
The pseudorotaxane formation behaviour of the viologen axle A1 and 24C8 was investigated by 1H NMR titration studies in CDCl3/CD3CN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The 1:1 complex, [2]pseudorotaxane A1·(24C8), was newly formed when 0.5 equiv. of 24C8 was added to the viologen axle A1, and the 1:2 complex, [3]pseudorotaxane A1·(24C8)2, was also observed upon further addition (1.0–5.0 equiv.) (Fig. 2a). The stepwise formation of A1·(24C8) and A1·(24C8)2 was also confirmed by ESI–TOF mass spectrometry (Fig. S1, ESI†). The binding constants, K1 and K2, for this two-step complexation (Fig. 1b) were determined by non-linear curve fitting of the changes in the mole fractions (Fig. 2a and Table 1). The negative cooperativity was clearly demonstrated (K1 = 1190 M−1, K2 = 132 M−1, and α = 4K2/K1 = 0.44), which can be explained by steric/electrostatic repulsion between the first and second wheels.16
:1). The 1:1 complex, [2]pseudorotaxane A1·(24C8), was newly formed when 0.5 equiv. of 24C8 was added to the viologen axle A1, and the 1:2 complex, [3]pseudorotaxane A1·(24C8)2, was also observed upon further addition (1.0–5.0 equiv.) (Fig. 2a). The stepwise formation of A1·(24C8) and A1·(24C8)2 was also confirmed by ESI–TOF mass spectrometry (Fig. S1, ESI†). The binding constants, K1 and K2, for this two-step complexation (Fig. 1b) were determined by non-linear curve fitting of the changes in the mole fractions (Fig. 2a and Table 1). The negative cooperativity was clearly demonstrated (K1 = 1190 M−1, K2 = 132 M−1, and α = 4K2/K1 = 0.44), which can be explained by steric/electrostatic repulsion between the first and second wheels.16
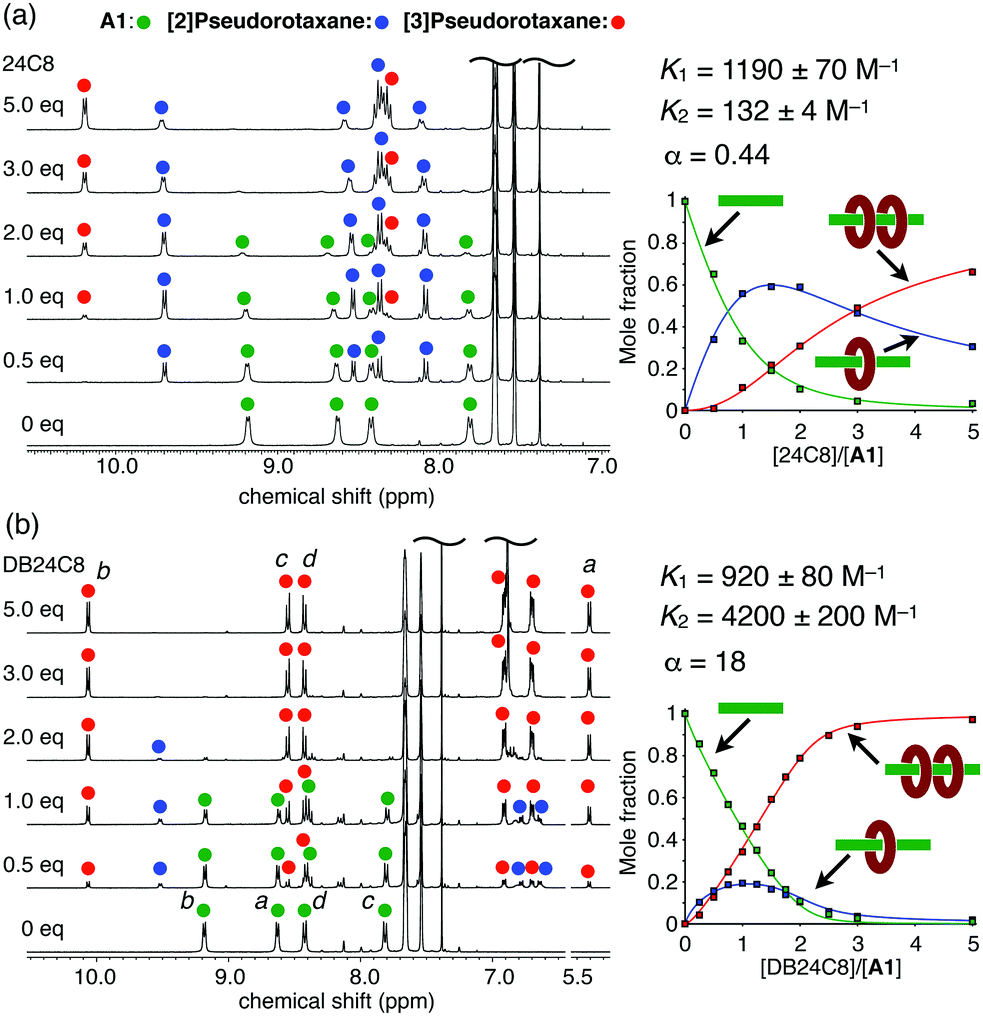 | ||
| Fig. 2
1H NMR spectral changes of A1 upon the addition of (a) 24C8 and (b) DB24C8 (400 MHz, 25 °C, CDCl3/CD3CN (4:1), [A1] = 5 mM). The plots of the mole fractions of the three components versus the equivalents of the wheel are also shown. See Fig. 1 for the signal assignments. | ||
| Axle | Crown ether | K 1 | K 2 | α |
|---|---|---|---|---|
|
a The binding constants were determined by 1H NMR spectroscopy in CDCl3/CD3CN (4:1) at 25 °C.
b Negligibly small.
|
||||
| A1 | 24C8 | 1190 ± 70 | 132 ± 4 | 0.44 |
| A1 | DB24C8 | 920 ± 80 | 4200 ± 200 | 18 |
| A1 | DN24C8 | 900 ± 500 | 4300 ± 1200 | ∼20 |
| A1 | DB30C10 | 1190 ± 190 | —b | ∼0 |
| A2 | DB24C8 | 36 ± 3 | —b | ∼0 |
| A2 | DN24C8 | 71 ± 6 | —b | ∼0 |
In contrast, a positive cooperativity was observed when DB24C8 was used instead of 24C8. Upon the addition of DB24C8 to A1, two sets of signals successively appeared in the 1H NMR spectra as observed in the A1-24C8 system (Fig. 2b), indicating the formation of the [2]- and [3]pseudorotaxanes (A1·(DB24C8) and A1·(DB24C8)2). The significant upfield shift of the viologen signal Ha upon the complexation (8.63 → 5.41 ppm) implied the strong shielding effect of the phenylene rings in DB24C8. In this case, 95% of A1 was converted into the [3]pseudorotaxane, A1·(DB24C8)2 when 3 equiv. of DB24C8 were added. This indicated that the DB24C8 more efficiently formed the [3]pseudorotaxane than 24C8, which was confirmed by the binding constants, K1 = 920 M−1 and K2 = 4200 M−1 (Fig. 2b and Table 1). To our surprise, this A1-DB24C8 system showed a highly positive cooperativity (α = 18), which is significantly greater than that of the A1-24C8 system (α = 0.44). A similar high cooperativity (K1 = 900 M−1, K2 = 4300 M−1, and α ≈ 20) was observed for the combination of A1 and DN24C8, which contains two naphthylene moieties (Fig. S5, ESI† and Table 1). Based on these results, we found that the introduction of aromatic rings into the 24C8 scaffold is essential to induce the positive cooperativity during the 1:2 complexation with A1.
In order to obtain further insight into the positive cooperativity, we investigated the complexation behaviour of A1 with the DB30C10 wheel, a larger macrocycle having two phenylene moieties. Upon the addition of DB30C10, the Ha signal in the viologen axle A1 was gradually shifted upfield, indicating the formation of the pseudorotaxane complex (Fig. S6, ESI†). The chemical shift changes were nicely fitted to the 1:1 binding isotherm with K1 = 1190 M−1 (Table 1). Thus, in this case, [2]pseudorotaxane A1·(DB30C10) was mainly formed and the formation of the [3]pseudorotaxane A1·(DB30C10)2 was negligible.17
We also investigated the complexation behaviour of the dibenzylviologen axle A2 in a similar manner. When DB24C8 or DN24C8 was added to A2, a new set of signals appeared in the 1H NMR spectra (Fig. S7 and S8, ESI†). The quantitative analysis yielded smaller binding constants for the 1:1 complex, K1 = 36 and 71 M−1 for DB24C8 and DN24C8, respectively, without forming the 1:2 complex (Table 1). Thus, the 1:2 complexes (e.g., [3]pseudorotaxanes) were formed only when the combination of diphenylviologen A1 and DB- or DN24C8 was used. This observation is consistent with previous reports describing that dialkyl- and dibenzylviologen axles form [2]pseudorotaxanes with DB24C8.11,12c
The formation of the [3]pseudorotaxane A1·(DB24C8)2 was significantly influenced by the solvent. The [3]pseudorotaxane formation became less efficient in more polar media containing less CDCl3 (Fig. S9–S13, ESI† and Table 2). Notably, however, higher cooperativity factors were observed in the polar solvents (α = 63 in CD3CN; α = 41 in CD3OD). Although the origin of the cooperativity is unclear at this point, the solvophobic effect or some other factors might contribute to the positive cooperativity.
| Solvent | K 1 | K 2 | α |
|---|---|---|---|
| a The binding constants were obtained by 1H NMR spectroscopy at 25 °C. b Negligibly small. | |||
| CDCl3/CD3CN (4:1) |
920 ± 80 | 4200 ± 200 | 18 |
| CD3CN | 48 ± 8 | 760 ± 90 | 63 |
| CDCl3/CD3OD (4:1) |
1500 ± 200 | 4800 ± 400 | 13 |
| CD3OD | 201 ± 13 | 2070 ± 70 | 41 |
| CDCl3/DMSO-d6 (4:1) |
420 ± 100 | 1500 ± 200 | 15 |
| DMSO-d6 | —b | —b | —b |
Single crystals of [3]pseudorotaxane A1·(DB24C8)2 were obtained by slow diffusion of pentane into a methanol/chloroform solution. As we expected, the A1 axle was threaded into the two DB24C8 molecules in the crystal structure (Fig. 3). The two DB24C8 molecules were arranged in an interdigitated fashion so as to wrap the two pyridinium rings, but no π–π stacking interaction was observed in the DB24C8–DB24C8 or DB24C8–A1 combinations (Fig. 3a and b). A similar interdigitated arrangement was found in the crystal structure of MV·(DB24C8)2, although the formation of the [3]pseudorotaxane was negligible in solution probably due to the lack of phenyl groups in the viologen axle.11 Each of the phenylpyridinium moieties was surrounded by eight oxygen atoms of the DB24C8 so that they can form eight intramolecular C–H⋯O hydrogen bonds ranging from 2.11 Å to 2.92 Å (Fig. 3c and Table S3, ESI†). This multiple hydrogen bonding, in which the phenyl C–H groups in A1 also participate, might account for the enhanced interaction between the A1 axle and the DB24C8 wheel. In addition, C–H⋯π interactions between the pyridinium C–H groups (Ha in Fig. 1a) in A1 and the phenylene rings in DB24C8 were also observed (from 2.66 Å to 2.93 Å, Fig. 3b). This interdigitated structure of [3]pseudorotaxane in solution was supported by the NOE correlation between Ha viologen proton and phenylene protons of DB24C8 (Fig. S14, ESI†).
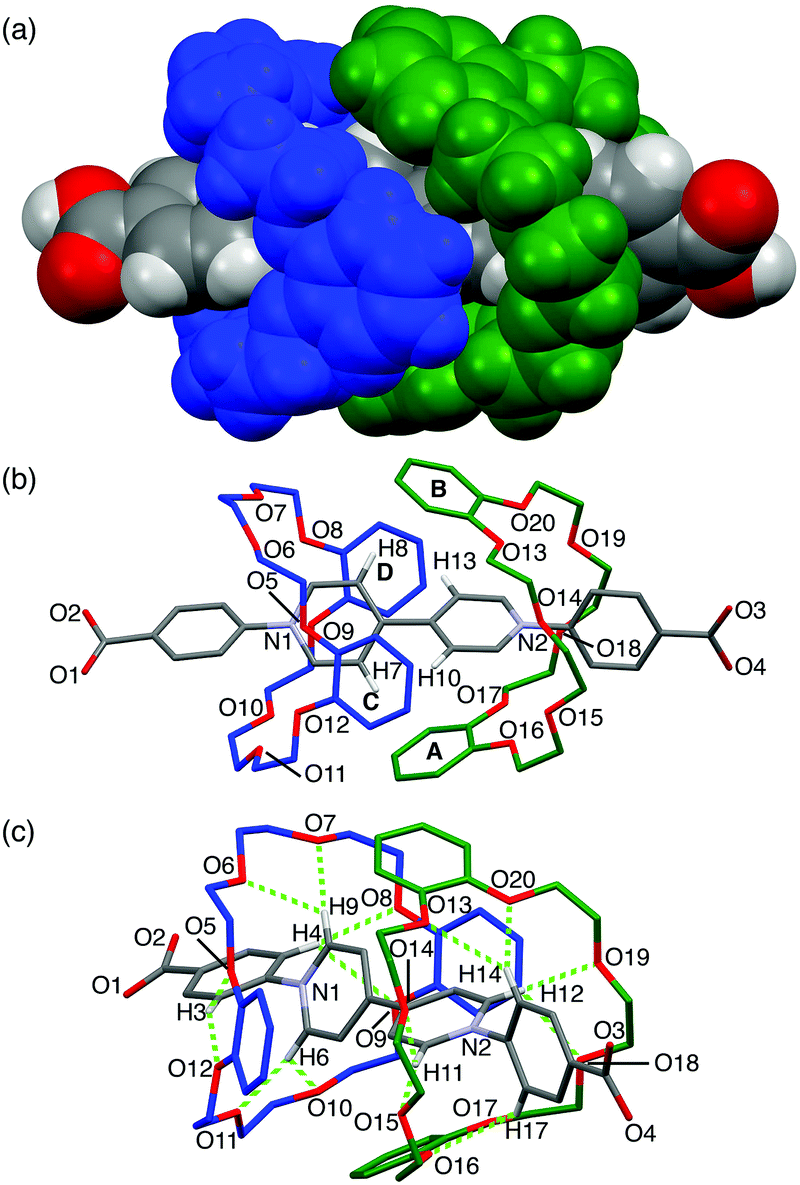 | ||
| Fig. 3 Crystal structure of A1·(DB24C8)2. (a) Space-filling model, and (b and c) capped stick models. Hydrogen atoms (b and c), solvent molecules and TFPB anions are omitted for clarity. Hydrogen atoms participating in the C–H⋯π and C–H⋯O interactions are shown in (b) and (c), respectively (C–H⋯π distances, H7⋯PhA, 2.66 Å; H8⋯PhB, 2.93 Å; H10⋯PhC, 2.74 Å; H13⋯PhD, 2.83 Å). The green dotted lines indicate the C–H⋯O hydrogen bonds (for the C–H⋯O distances, see Table S3, ESI†). | ||
Finally, we applied this positive-cooperative binding to the efficient synthesis of [3]rotaxane by end-capping of the carboxy groups in A1via amide formation (Fig. 4a).18 We prepared a solution containing A1 (20 mM) and DB24C8 (40 mM) in CDCl3/CD3CN (4:1), in which 89% of A1 is converted to [3]psedorotaxane A1·(DB24C8)2 as estimated from the binding constants. The end-capping was achieved without dissociation of the wheel components by amide formation with 3,5-di-tert-butylaniline under the conditions using 1-hydroxybenzotriazole (HOBt) and 1-ethyl-3-(3-dimethylamino)propylcarbodiimide (EDC). The almost exclusive formation of [3]rotaxane R1·(DB24C8)2 was confirmed by the 1H NMR spectrum (Fig. 4b) and the ESI-TOF mass spectrum (m/z = 834.9 for [R1·(DB24C8)2 − 2TFPB]2+, Fig. S15, ESI†). Notably, the signals for the [2]rotaxane R1·(DB24C8) were scarcely observed in the 1H NMR spectrum even though only a stoichiometric amount of the DB24C8 wheel was used. Thus, the [3]rotaxane R1·(DB24C8)2 was efficiently synthesized without consuming an excess amount of the wheel component based on the newly discovered [3]pseudorotaxane system exhibiting a positive-cooperative complexation behaviour.
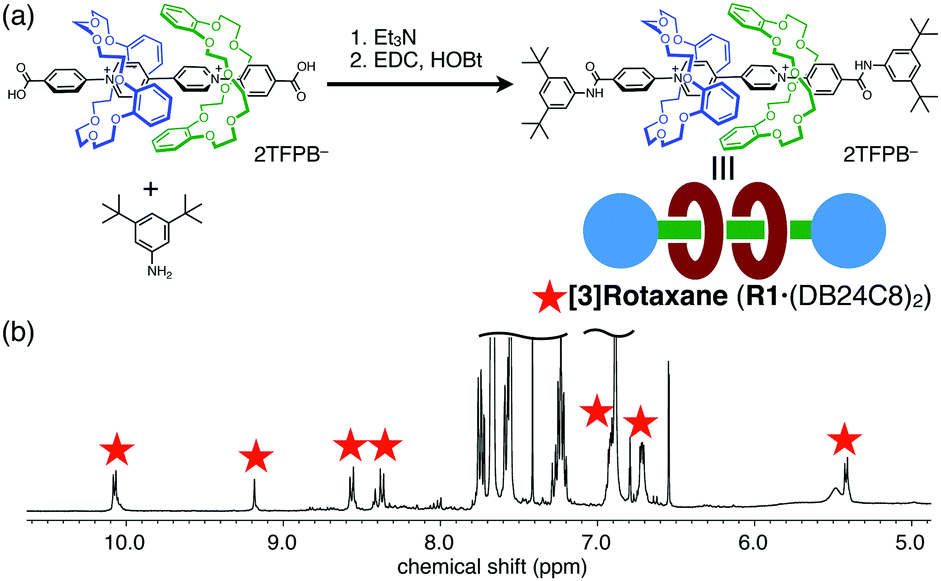 | ||
| Fig. 4 (a) Reaction scheme of [3]rotaxane R1·(DB24C8)2 formation by amide formation, and (b) 1H NMR spectrum of the reaction mixture (400 MHz, CDCl3/CD3CN (4:1)). | ||
In conclusion, we have demonstrated a new positive-cooperative [3]pseudorotaxane system consisting of DB24C8 and the diphenylviologen axle A1. The maximum cooperativity factor of 63 was achieved in the CD3CN solution. The X-ray crystallographic analysis revealed that both the multiple C–H⋯O and C–H⋯π interactions between the A1 and DB24C8 components contributed to the stabilization of the [3]pseudorotaxane. With these results in hand, [3]rotaxane R1·(DB24C8)2 was efficiently obtained via the amide formation from a 1:2 stoichiometric mixture of A1 and DB24C8. We believe that these results open the way to the development of not only new [3]rotaxanes based on various kinds of DB24C8 derivatives, but also oligo- and polyrotaxanes that have more than two DB24C8 wheels for potential application to more complex molecular machines. The investigation of the electrochemical responses of the [3]rotaxane based on the redox active viologen unit is currently underway.
This work was supported by JSPS KAKENHI Grant Number JP20H04667, JP18H04511 (Soft Crystals), and JP16H06510 (Coordination Asymmetry), Inoue Science Research Award, the Asahi Glass Foundation, the Iwatani Naoji Foundation, and the World Premier International Research Center Initiative (WPI), MEXT, Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, New Jersey, 2016 CrossRef; (b) G. Schill, Catenanes, Rotaxanes, and Knots, Academic Press, New York, 1971 Search PubMed; (c) D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS; (d) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; (e) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed; (f) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- (a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart., Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (c) P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed; (d) R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS PubMed.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- (a) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed; (b) J.-P. Collin, J. Frey, V. Heitz, J.-P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609–5620 CrossRef CAS PubMed; (c) H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 CrossRef PubMed.
- (a) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS; (b) R. S. Forgen, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef PubMed; (c) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- (a) P. R. Ashton, M. Bělohradský, D. Philp, N. Spencer and J. F. Stoddart, J. Chem. Soc., Chem. Commun., 1993, 1274–1277 RSC; (b) D. B. Amabilino, P. R. Ashton, M. Bělohradský, F. M. Raymo and J. F. Stoddart, J. Chem. Soc., Chem. Commun., 1995, 747–750 RSC; (c) N. Watanabe, T. Yagi, N. Kihara and T. Takata, Chem. Commun., 2002, 2720–2721 RSC; (d) H. W. Gibson, N. Yamaguchi and J. W. Jones, J. Am. Chem. Soc., 2003, 125, 3522–3533 CrossRef CAS PubMed; (e) E. A. Neal and S. M. Goldup, Chem. Sci., 2015, 6, 2398–2404 RSC.
- (a) L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC; (b) M. E. Belowich, C. Valente, R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243–5261 CrossRef CAS PubMed.
- F. Huang, F. R. Fronczek and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 9272–9273 CrossRef CAS PubMed.
- (a) S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2003, 5, 704–710 CrossRef PubMed; (b) Z. Peng, X. Guo, W. Xu, J. Li, P. Deng, X. Xiao, W. Feng and L. Yuan, Chem. Commun., 2019, 55, 4869–4872 RSC; (c) Z. Ye, J. Wang, S. S. K. Kothapalli, Z. Yang, L. Chen, W. Xu, Y. Cai, T. Zhang, X. Xiao, P. Deng, W. Feng and L. Yuan, Chem. Commun., 2020, 56, 1066–1069 RSC; (d) C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed; (e) C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS PubMed; (f) M. V. Rekharsky, H. Yamamura, M. Kawai, I. Osaka, R. Arakawa, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Org. Lett., 2006, 8, 815–818 CrossRef CAS PubMed; (g) Y.-L. Ma, H. Ke, A. Valkonen, K. Rissanen and W. Jiang, Angew. Chem., Int. Ed., 2018, 57, 709–713 CrossRef CAS PubMed; (h) L.-S. Zheng, J.-S. Cui and W. Jiang, Angew. Chem., Int. Ed., 2019, 58, 15136–15141 CrossRef CAS PubMed; (i) Q. Wang, Y. Zhong, D. P. Miller, X. Lu, Q. Tang, Z.-L. Lu, E. Zurek, R. Liu and B. Gong, J. Am. Chem. Soc., 2020, 142, 2915–2924 CrossRef CAS PubMed.
- K. Nikitin and H. Müller-Bunz, New J. Chem., 2009, 33, 2472–2478 RSC.
- (a) B. L. Allwood, N. Spencer, H. Shahriari-Zavareh, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1064–1066 RSC; (b) P. R. Ashton, D. Philp, N. Spencer and J. F. Stoddart, J. Chem. Soc., Chem. Commun., 1992, 1124–1128 RSC; (c) A. B. Braunschweig, C. M. Ronconi, J.-Y. Han, F. Aricó, S. J. Cantrill, J. F. Stoddart, S. I. Khan, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 2006, 1857–1866 CrossRef CAS; (d) H. R. Wessels, C. Slebodnick and H. W. Gibson, J. Am. Chem. Soc., 2018, 140, 7358–7370 CrossRef CAS PubMed; (e) T. B. Gasa, J. M. Spruell, W. R. Dichtel, T. J. Sørensen, D. Philp, J. F. Stoddart and P. Kuzmič, Chem. – Eur. J., 2009, 15, 106–116 CrossRef CAS PubMed.
- H. Nishida, N. Takada, M. Yoshimura, T. Sonoda and H. Kobayashi, Bull. Chem. Soc. Jpn., 1984, 57, 2600–2604 CrossRef CAS.
- J.-J. Liu, Y.-F. Guan, M.-J. Lin, C.-C. Huang and W.-X. Dai, Cryst. Growth Des., 2015, 15, 5040–5046 CrossRef CAS.
- M. R. Geraskina, A. S. Dutton, M. J. Juetten, S. A. Wood and A. H. Winter, Angew. Chem., Int. Ed., 2017, 56, 9435–9439 CrossRef CAS PubMed.
- K. A. Connors and D. D. Pendergast, J. Am. Chem. Soc., 1984, 106, 7607–7614 CrossRef CAS.
- For [2]rotaxane containing DB30C10 and viologen derivative, see ref. 12d.
- M.-J. Blanco, J.-C. Chambron, V. Heitz and J. P. Sauvage, Org. Lett., 2000, 2, 3051–3054 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, crystallographic data, additional 1H NMR spectra and ESI-TOF MS for the complexes. CCDC 1998063. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03131c |
| This journal is © The Royal Society of Chemistry 2020 |