
Operando vibrational spectroscopy for electrochemical biomass valorization
Nina
Heidary
and
Nikolay
Kornienko
 *
*
Department of Chemistry, Université de Montréal, Roger-Gaudry Building, Montreal, Quebec H3C 3J7, Canada. E-mail: nikolay.kornienko@umontreal.ca
First published on 13th May 2020
Abstract
Electrocatalysis is a promising route to generate fuels and value-added chemicals from abundant feedstocks powered by renewable electricity. The field of electrocatalysis research has made great progress in supplementing electrocatalyst development with operando vibrational spectroscopic techniques, those carried out simultaneously as the reaction is occurring. Such experiments unveil reaction mechanisms, structure–activity relationships and consequently, accelerate the development of next generation electrocatalytic systems. While operando techniques have now been extensively applied to water electrolysis and CO2 reduction, their application to the emerging area of biomass valorization is rather nascent. The electrocatalytic conversion of biomass can provide an alternate, environmentally friendly route to the chemicals which power our society, but this field still requires much growth before the envisioned technologies are economically competetive with thermochemical routes. Within this context, a growing body of work has begun to translate the methodology and concepts from water/CO2 electrolysis to biomass valorization to elucidate links between catalyst structure, adsorbed surface intermediates, and the resultant catalytic performance. The reactions of interest here include the upgrading of biomass platforms such a 5-hydroxymethylfurfural or glycerol to value-added chemicals. In this feature article we highlight these efforts and provide a critical view on the steps necessary to take to further progress the field. We further show how the knowledge derived from these studies can be translated to a plethora of other organic transformations to forge new avenues in renewable energy electrocatalysis.
Renewable energy electrocatalysis
As the consequences of excessive fossil fuel consumption become increasingly prevalent,1 efforts to transition to a sustainable society are critical.2 While progress is certainly being made, more research into the utilization of renewable energy as a source for the fuels and chemical feedstocks that power our lives is necessary.Within this context, electrocatalysis stands to play an ever more important role.3 There exist many mature technologies that can convert renewable energy sources (sunlight, hydro, wind…) into electricity. However, the intermittency of renewable sources and society's need for energy dense fuels and chemical feedstocks requires that the generated electricity be converted to an energy-dense storable medium. To this end, both industry and academia are heavily investigating electrocatalysts that generate H2 from water as an energy carrier and carbon-based fuels and value-added chemicals from CO2. There has been much emphasis placed on these reactions over the last decade, and a diverse array of research in catalyst design, reactor engineering, theoretical modelling and mechanistic investigations.4–6 As a result, these technologies have matured to the point where they are potentially cost-competitive with fossil-fuel based pathways.7,8
Moving beyond water electrolysis and CO2 reduction, researchers within the field of electrocatalysis have recently placed emphasis on electrocatalytic reactions that convert waste biomass and biomass platforms9 to value-added chemicals.10,11 This approach generates the products that power the chemical industry via a renewable-energy driven process, replacing the energy-intensive, fossil fuel-derived thermochemical routes from which they are currently generated (Fig. 1). The electrochemical valorization of compounds such as glycerol,12–14 furfural,15,16 5-hydroxymethylfurfural (HMF)17,18 can (1) improve the economics19 of biofuels processes (e.g. making use of glycerol byproducts from biodiesel production) and (2) in many cases these reactions can be inserted into existing electrolysis systems,20–23 (e.g. replacing water oxidation with biomass oxidation at the anode of an electrolyser) to decrease the total energy requirement and co-generate valuable products.
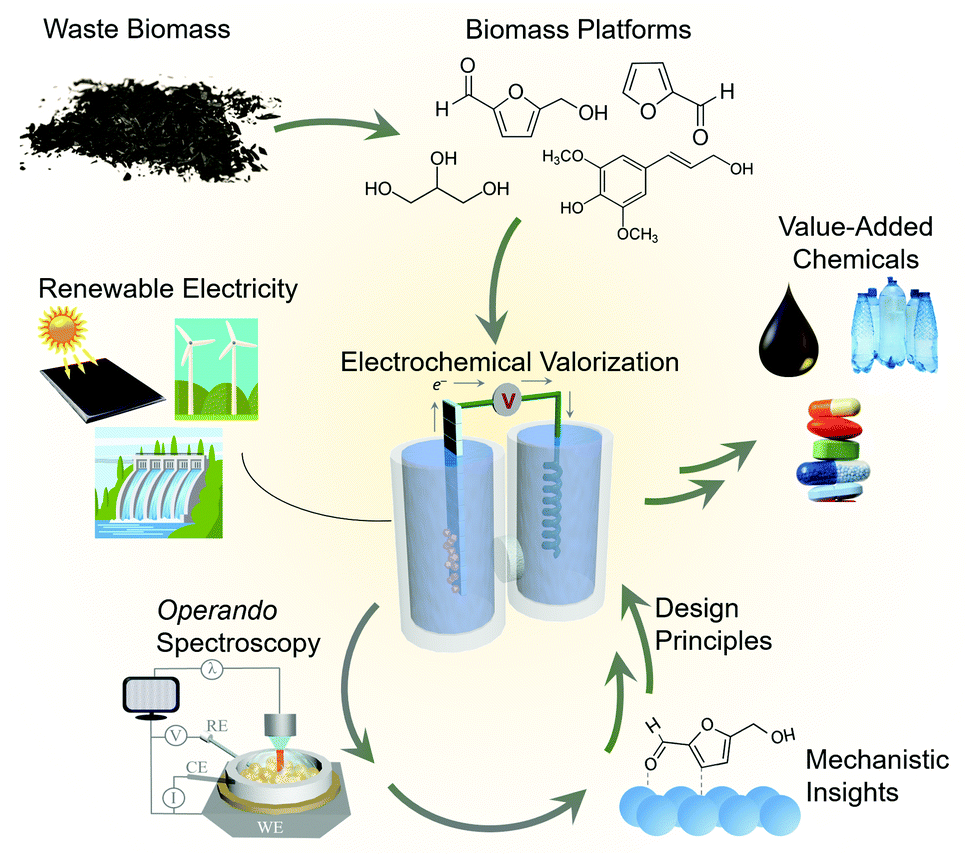 | ||
| Fig. 1 The role of operando spectroscopy in the electrochemical biomass valorization process. | ||
Mechanistic insights into biomass valorization
While electrochemical biomass valorization is certainly appealing from both an environmental and economic aspect, the field is not yet mature and there is much room to grow in developing electrocatalysts that are cost-effective, efficient, and selective for a desired product.24 Like in the aforementioned cases of water and CO2 electrolysis, mechanistic investigations into catalyst structure–activity relationships and surface reaction pathways are anticipated to play an important role in the field's development (Fig. 1). Pertinent questions to be answered are (1) what are the surface reaction intermediates, both under steady state and dynamic conditions, (2) what are the surface reaction sites of the catalyst and which types of sites are the most active or selective, and (3) how do electrolyte and secondary coordination sphere effects modulate the reaction landscape as previously investigated for CO2 reduction,25,26 for example. Here, both the knowledge base and developed methodology can be used as a blueprint to facilitate the experimental design, data interpretation, and catalyst development. Within this context, this feature article focuses on recent investigations that utilize operando techniques, those that are performed simultaneously as an electrocatalytic system is operating, to extract insights into electrochemical biomass valorization. In many cases, similar setups (e.g. cell design, spectrometer configuration…) as for water/CO2 electrolysis can be employed and an analogous approach to data interpretation can be followed.27–30 Vibrational spectroscopy has been the most widely used approach to date here and will consequently be the focus of this feature article. We do not aim to provide an exhaustive list of works in which operando spectroscopy was used to probe biomass oxidation, rather we wish to highlight several works to provide an insights and perspectives into this emerging area.Raman spectroscopy
Raman spectroscopy probes inelastically scattered radiation upon irradiating a sample with a monochromatic light source as its various vibrational modes are excited.31 Raman spectroscopy can therefore provide information on both the molecular intermediates of a reaction and on the catalyst structure.32 Furthermore, because scattered radiation is collected, fairly simple reaction setups can be utilized to integrate Raman spectroscopy with an electrochemical cell (Fig. 2a).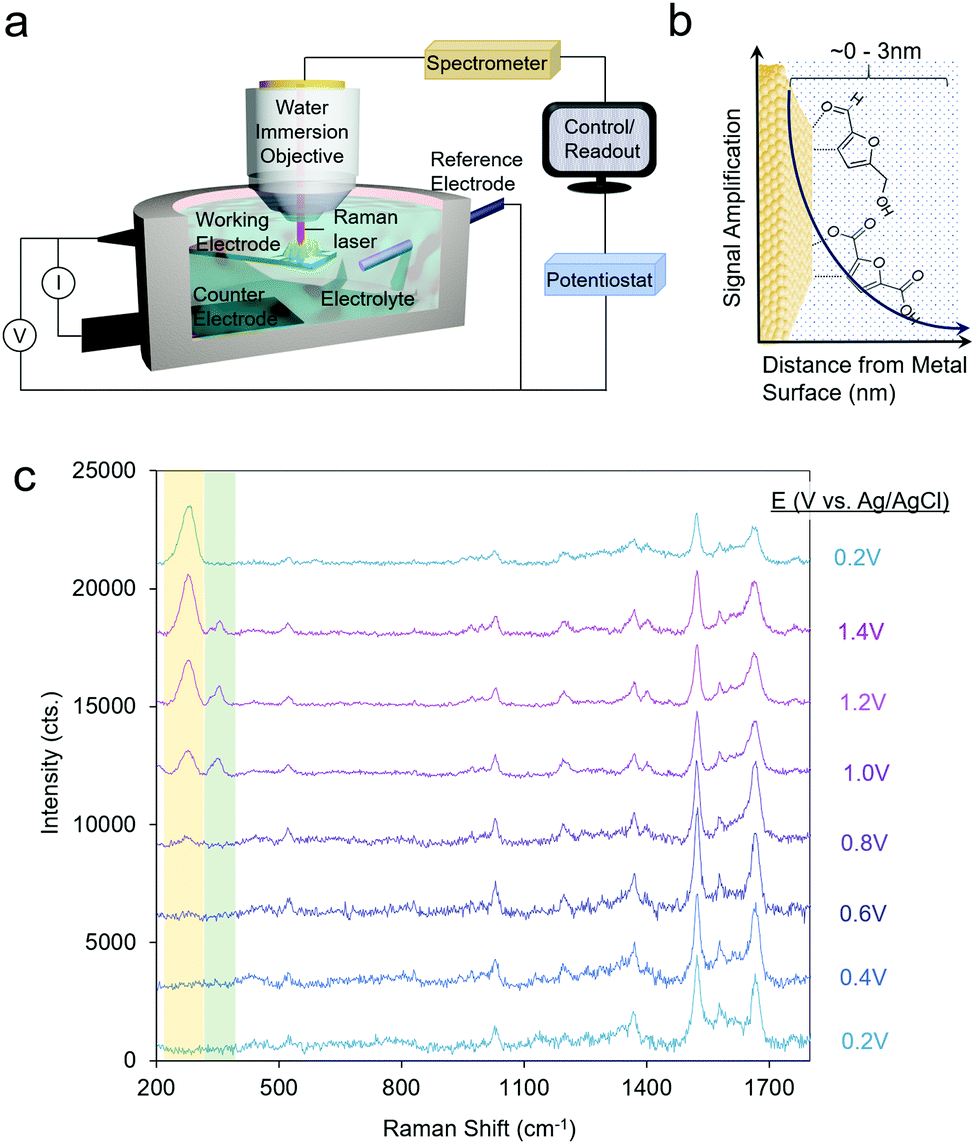 | ||
| Fig. 2 A typical reaction cell for operando SERS measurements (a), which investigate species precisely at the catalyst–electrolyte interface (b). SERS spectra of HMF oxidation on Au. Reprinted from ref. 28 (N. Heidary and N. Kornienko, Chem. Commun., 2019, 55, 11996–11999). Copyright 2019 Royal Society of Chemistry. | ||
A significant drawback to this technique is that only a small fraction of the incident radiation is scattered (on the order of one out of ∼106 photons) and consequently, the sensitivity is inherently low. To overcome this, surface-enhanced Raman spectroscopy (SERS) is often utilized, which takes advantage of the signal enhancement conferred by plasmonic substrates.33 Briefly, the optical excitation of plasmonic nanomaterials (e.g. Ag and Ag) with a laser which matches the resonance frequency of their valence electrons leads to enhanced electromagnetic fields at the surface and consequently a rise in the Raman signal, which arises from vibrational modes which exhibit changes in polarizability. A convenient advantage to SERS is that the signal enhancement arises from species only within the first few nm of the plasmonic particle surface (Fig. 2b). This renders SERS especially surface-selective and allows researchers to focus on only the catalyst–electrolyte interface without interference from the bulk solution.
Operando SERS experiments were recently undertaken to investigate the oxidation of HMF on gold.34 In a 10 mM KOH electrolyte, furan-2,5-dicarbaldehyde (DFF) was the primary product, arising from the oxidation of the alcohol group on HMF. SERS measurements revealed that HMF adsorbed by both its furan ring and alcohol group and a surface-bound alkoxy intermediate was present at the oxidizing potentials in which the reaction occurred. Interestingly, at least 2 types or surface-active sites were seen with likely different reactivity, as evidenced through two separate bands at 335 and 351 cm−1, which progressively rose and fell in intensity as the potential of the electrode was systematically shifted in the positive (oxidizing) direction (Fig. 2c). This study marked the first example of SERS utilized to investigate electrochemical HMF oxidation.
This line of investigations was subsequently extended to study transition metal oxides, which are more active than Au for HMF oxidation. However, because these materials do not give rise to a SERS enhancement, a system was designed which incorporated a plasmonic core and a thin catalytic overlayer (Fig. 3a).35 Electrodeposited Au dendrites were covered with a thin, ∼1–2 nm layer of the desired transition metal which was coated overtop through an electrodeposition and galvanic displacement process. Importantly, this system allowed for the investigation of both the reaction intermediates as well as the changes occurring within the catalyst under an applied electrochemical bias (Fig. 3b and c).
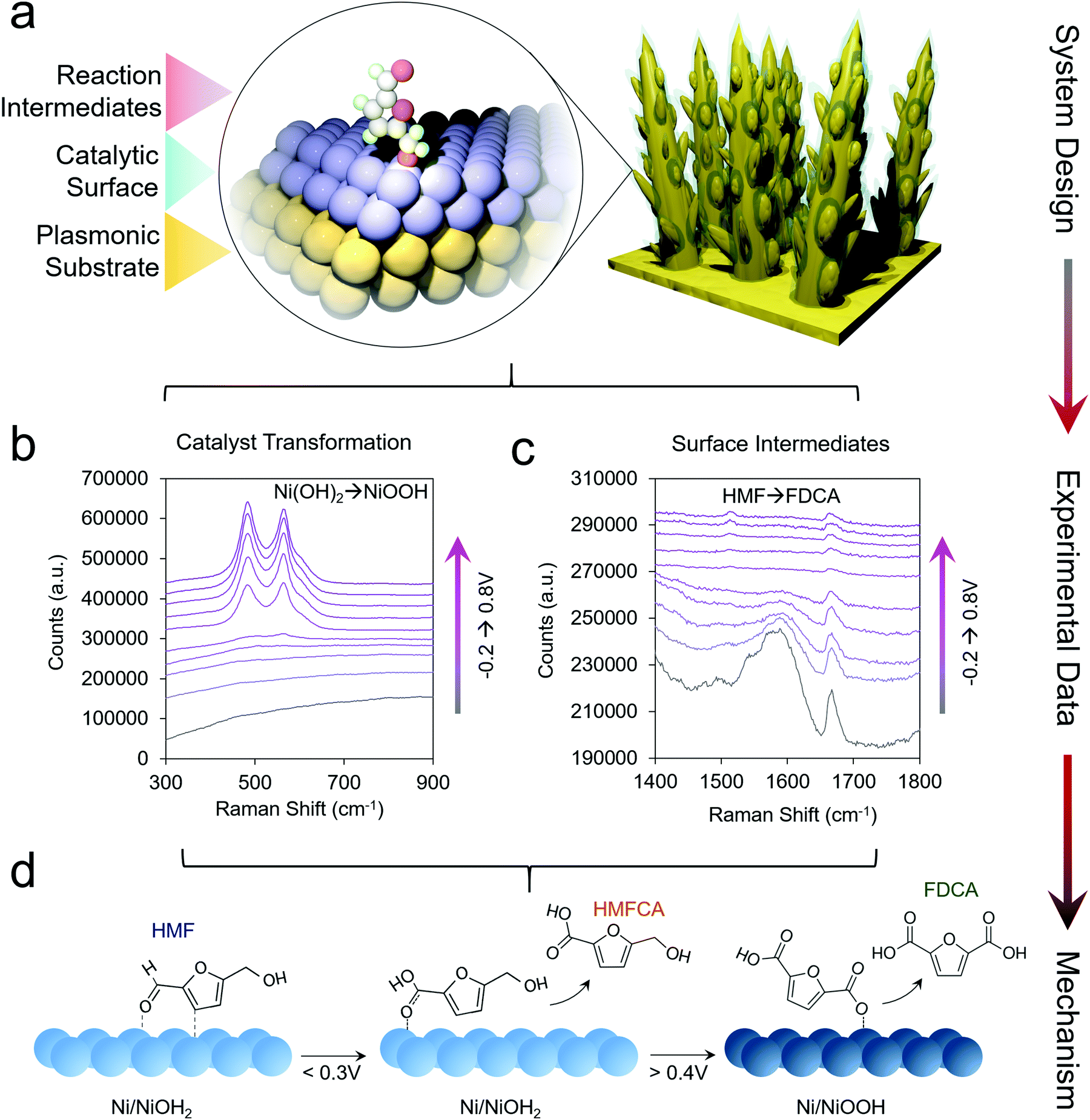 | ||
| Fig. 3 Catalyst materials are designed for utilizing SERS to investigate non-plasmonic surfaces (a). Here, the catalyst transformation (b) and surface intermediates (c) can be probed by SERS to generate a series of mechanistic insights enabling the rational design of next-generation systems (d). Reprinted with permission from ref. 29 (N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806). Copyright 2020 Royal Society of Chemistry. | ||
Using Ni as the initial catalytic material, which spontaneously forms a NiOH2 surface layer in alkaline conditions, HMF oxidation was investigated in 10 mM and 1.0 M KOH. In 10 mM KOH, DFF was the main product and HMF was believed to interact with the NiOH2 surface through its alcohol group. Interestingly, in 10 mM KOH, the NiOOH phase readily formed under oxidizing conditions in the absence of HMF, as evidenced by two strong bands at 490 and 561 cm−1. However, when 10 mM HMF was added to the solution, NiOOH was only seen in very small amounts, presumably because the strong binding of HMF to the Ni(OH)2 surfaces hindered its formation. Furthermore, the Ni(OH)2 catalyst did not interact strongly with the aldehyde group of the reactants and the reaction consequently stopped at DFF, instead of proceeding towards the desired 2,5-furandicarboxylic acid (FDCA) product, which required the oxidation of both alcohol and aldehyde groups.
In 1.0 M KOH, NiOOH did indeed form, and under these conditions the catalyst was highly active for both alcohol and aldehyde oxidation, generating FDCA with close to 100% selectivity (Fig. 3d). The reaction proceeded exceptionally fast once NiOOH was generated, pointing to the importance of this species in the catalytic cycle. Beyond the identification of the NiOOH formation, SERS measurements provided evidence for voltage-dependent, oxygen-bound surface intermediates en route to FDCA formation (Fig. 3d). Oxides of Co, Fe, CoFe and NiFe were also investigated and interestingly, the most active water oxidation catalysts, NiFe and CoFe oxides, were not the most active HMF oxidation catalysts. While initial structure–activity relationships were established in this work, many questions still remain. The precise effects due to defects, strain, grain boundaries and undercoordinated catalytic sites of HMF oxidation catalysts are still to be determined. The effects of electrolyte and double-layer composition are also not fully clarified.
Operando Raman investigations were also applied to the oxidation of glycerol on CuO surfaces.36 While there was no SERS enhancement, enough products built up over the course of an electrolysis experiment to measure the changes in the bulk electrolyte solution over time. The authors detected bands in the spectra corresponding to formate, glyceraldehyde (GALD), dihydroxyacetone (DHA), and glycolate over the time course of one hour. The Raman data, in combination with that obtained from bulk product analysis, helped the authors put forth a mechanistic pathway in which glycerol can be selectively (60% selectivity) converted at moderate pH values (pH 9) but in alkaline solutions of pH 13, DHA converts in solution to GALD, and this is further oxidized to glycolate, formate, oxalate and carbonate.
A promising approach towards further SERS-driven investigation of HMF and glycerol oxidation on a multitude of non-plasmonic catalytic systems would be to employ shell-isolated nanoparticles (SHINs) (Fig. 4).37 Noble metal nanoparticles, covered with a thin shell of an inert material (e.g. alumina, silica or zirconia), that sit overtop of a catalyst material, would also function to confer a SERS enhancement without the stringent requirement of having a nm-thick catalyst overlayer. This technique, for example, helped elucidate the reaction mechanisms of oxygen reduction on single crystal platinum surfaces, with the aid of Au@SiO2 SHINS.38
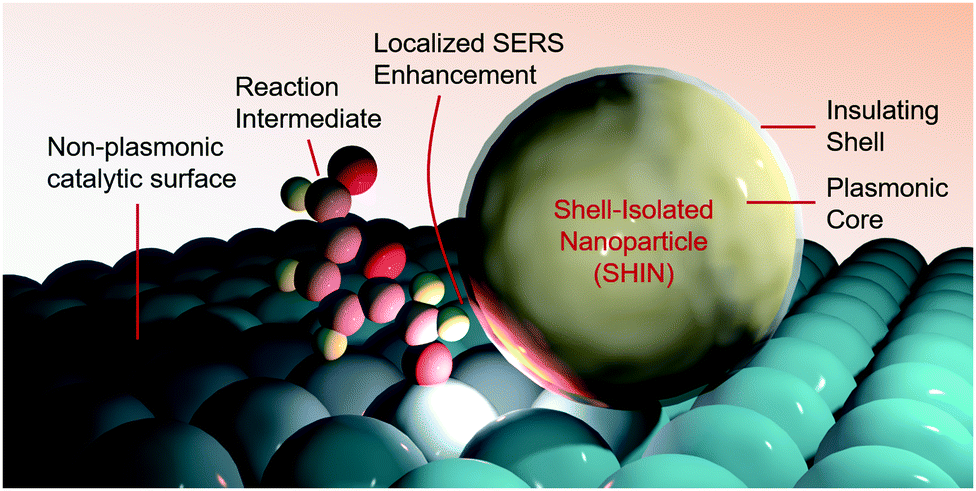 | ||
| Fig. 4 Shell-isolated nanoparticles function to enable SERS measurements on non-plasmonic catalyst surfaces. | ||
Infrared spectroscopy
As opposed to Raman spectroscopy, which detects vibrational modes exhibiting a change in polarizability, infrared (IR) spectroscopy probes vibrational modes that feature a change in their dipole moment. Translating IR to use as an operando technique to investigate electrocatalytic processes is often done in attenuated total reflection (ATR) mode. In this configuration, an ATR waveguide is coated with a catalyst layer that is electrically connected and functions as the working electrode. The IR light is coupled from below, and the evanescent IR beam penetrates ∼1 μm into the solution, depending on the waveguide's material (Fig. 5a and b). Much like SERS, the ATR-IR signals can also be enhanced through the use of plasmonic substrates in what is called surface-enhanced IR spectroscopy (SEIRAS).39,40 Here, the penetration depth is also on the order of several nm, increasing the surface-sensitivity of the measurement (Fig. 5b).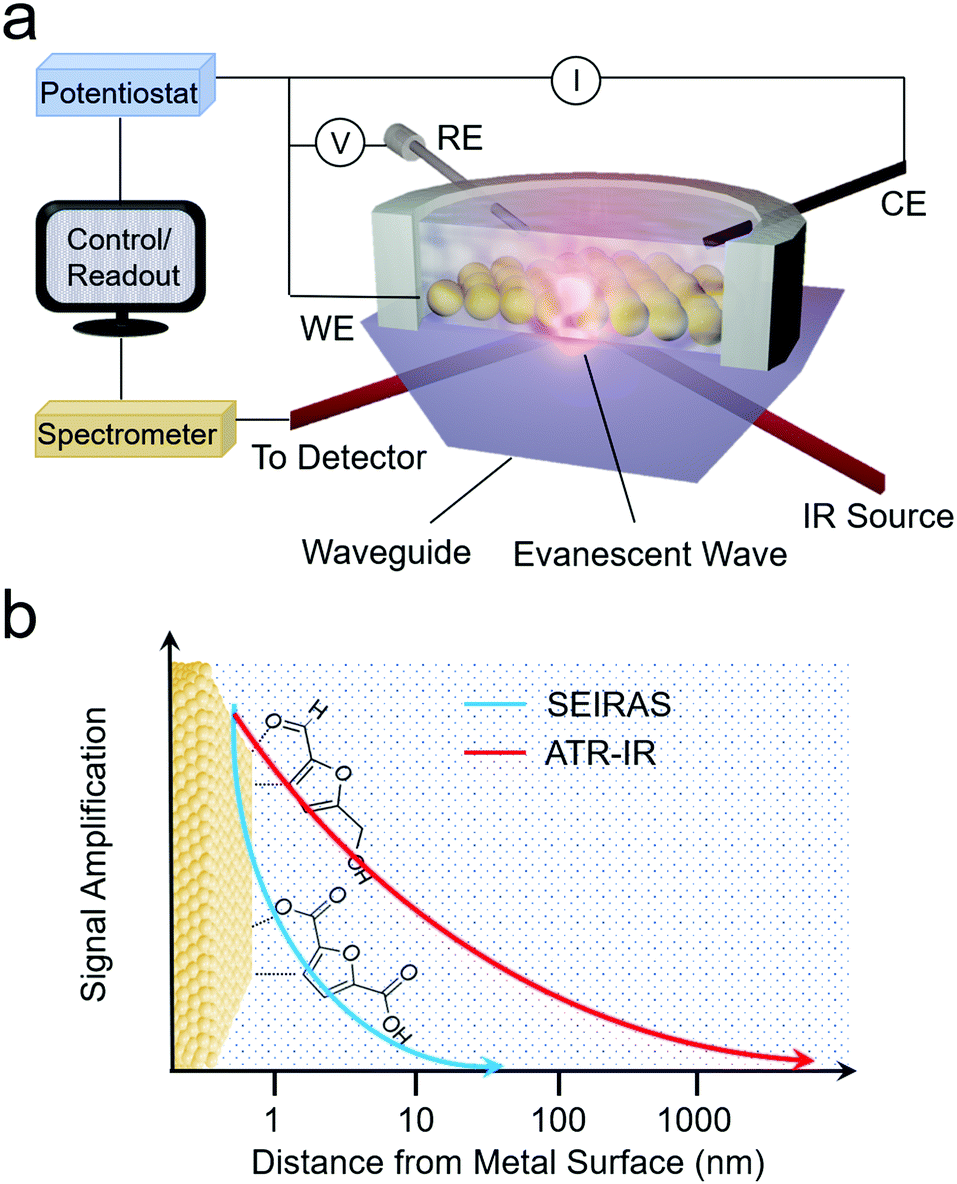 | ||
| Fig. 5 Schematic of ARE-IR spectroelectrochemical setup (a) and signal enhancement as a function of distance from the electrode surface for ATR-IR and SEIRAS techniques (b). | ||
Operando ATR-IR spectroscopy was recently applied to probe electrochemical HMF oxidation on nickel boride catalysts.41 Upon the application of an increasingly oxidizing voltage, the authors observed bands first for 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) then bands corresponding to 5-formyl-2-furancarboxylic acid (FFCA) and finally bands indicating the presence of FDCA. This experiment, coupled to bulk product analysis, indicated that HMF oxidation in this system proceeds first via the oxidation of the aldehyde group rather than the alcohol group.
ATR-IR has also been applied to investigate the oxidation of glycerol on a number of different catalytic surfaces. A notable study utilized ATR-IR to probe the reaction mechanism and thus bring forth structure–activity relationships in a series of compositionally variant, porous PdBi bimetallic catalysts.42 At weakly oxidizing potentials, the authors detected GALD and DHA as the primary intermediates. As the potential was poised more and more oxidizing, the hydroxypyruvate intermediate was detected and finally CO2 as complete oxidation of glycerol occurred and its C–C bonds were cleaved. Pd was hypothesized to serve as the active site while the role of Bi was to protect the Pd from oxidation, increasing both activity and stability of the material. The Pd atoms adjacent to Bi were also postulated to feature differing behaviour in terms of the reactants that they adsorbed, which would explain the differing selectivity of PdBi and Pd. As an example of this, the hydroxypyruvate intermediate detected via operando ATR-IR only on PdBi, was proposed to be facilitated by the addition of *OH species which were present at Pd sites adjacent to a Bi atom.
Pt surfaces covered with sub-monolayers of Bi exhibited differing glycerol oxidation activity as compared with bare Pt.43 Bi increased both the activity of Pt(111) and the selectivity for DHA. Bands attributed to the oxidation products of formic acid, carbon monoxide and carbon dioxide were detected.43 The spectra and therefore surface reaction pathway closely matched that from pure Pt (Fig. 6a and b),44 and from this the authors inferred that the presence of Bi does not change the reaction pathway. Instead, the Bi stabilizes the enediol intermediate, the key species prior to GALD or DHA formation (Fig. 6c).
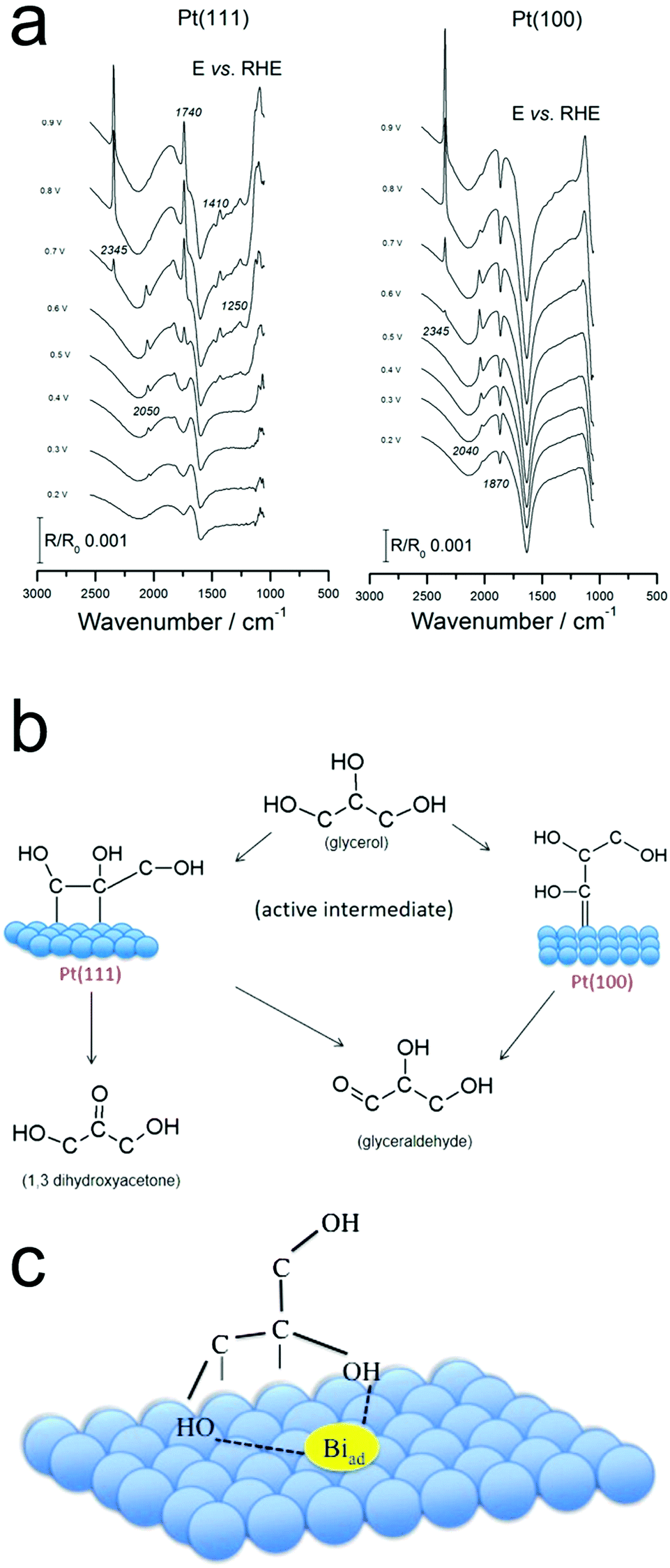 | ||
| Fig. 6 Operando IR spectra on Pt (a) point to the differing reaction pathways on Pt(111) and Pt(100) (b). The presence of surface-bound Bi is thought to stabilize the enediol intermediate (c). Reprinted with permission from ref. 37 (A. C. Garcia, Y. Y. Birdja, G. Tremiliosi-Filho and M. T. M. Koper, J. Catal., 2017, 346, 117–124), ref. 38 (A. C. Garcia, M. J. Kolb, C. van Nierop y Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, ACS Catal., 2016, 6, 4491–4500). Copyright 2017 Elsevier, 2016 American Chemical Society. | ||
The method of PdAu catalyst preparation was found to play a significant role in their glycerol oxidation activity in an alkaline 0.1 M NaOH electrolyte.45 Catalysts prepared by immersing a Au electrode in a PdSO4 solution were found to be the most active towards the complete oxidation of glycerol to carbonate. Operando ATR-IR measurements illustrated the difference in mechanism that the Pd overlayer imparts. Surface adsorbed CO, stemming from the oxidative cleavage of CH2OH fragments, was only found in the presence of Pd. Furthermore, the PdAu catalysts gave rise to a strong band attributed to CO2 near the interface, and thus hinted at a depressed local pH at the surface. Interestingly, the potential-dependence of CO and CO2 did not match and from this observation, the authors speculated that CO2 does not originate from a CO precursor but rather from an alternate species such as formate. From the sum of the data, the bimetallic catalysts’ high activity was attributed to the synergistic function of the Pd, which was more active to C–C bond cleavage, and Au, which oxidized the fragments to CO2 and carbonate end products.
Pt, Pd and Au catalysts were compared for glycerol oxidation in 1.0 M NaOH with IR reflectance.46 Each catalyst featured similar absorption bands under catalytic conditions indicating a similar surface reaction pathway, with one primary exception: surface bound CO was only detected on Pt and Pd, stemming from a cleavage of C–C bonds at low potentials from a dissociative adsorption of glycerol on Pt and Pd. However, the CO species is very transient on Au and may have rapidly desorbed. Furthermore, hydropyruvate was spectroscopically detected only on Au.
Furfural oxidation in 0.25 M HClO4 on polycrystalline Pt surfaces was recently investigated with a combination of online electrochemical mass spectrometry (OLEMS) and ATR-SEIRAS.47 At low potentials of 0.9–1.0 V vs. RHE, 2-furoic acid (FA) was the primary product. At more positive potentials, 2(3H)-furanone, maleic acid, 5-hydroxyfuroic acid, and 5-hydroxy-2(5H)-furanone increasingly formed. SEIRAS spectra were acquired during a slow cyclic voltammogram of furfural and of FA, as a potential intermediate to further oxidized products. At low potentials (0.3 V vs. RHE) linearly-bound and bridge-bound CO was detected as a product of decarbonylation of furfural and FA (Fig. 7a and b). The CO remained bound to the surface until the Pt oxidized at 1.0 V. Once the Pt oxidized, additional intermediates could be detected and were tentatively attributed to 2(5H)-furanone and other surface bound furanone species. Further, high electrochemical oxidation currents were only attained when the surface was cleared of CO as this species was thought to poison the surface and block further reactivity until it could be oxidized off. The selectivity to C4 and C5 species over CO2 was attributed to a continual steady state coverage of organic intermediates. From a combination of their experimental data, the authors proposed a series of pathways resulting in their observed product distribution (Fig. 7c).
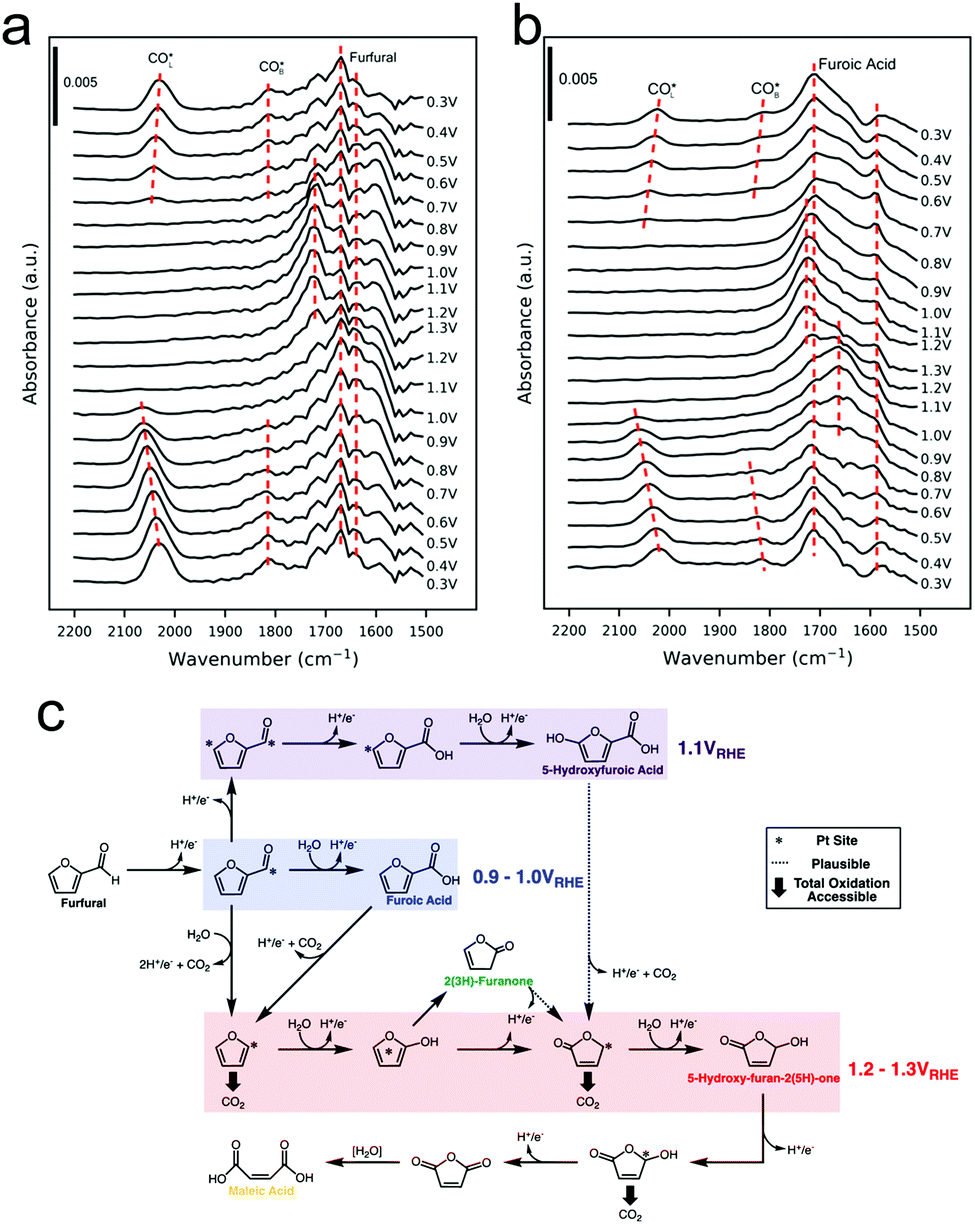 | ||
| Fig. 7 SEIRAS spectra of furfural (a) and furoic acid (b) oxidation on polycrystalline Pt. A series of pathways from the experimental data was proposed (c). Reprinted with permission from ref. 41 (A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316). Copyright 2019 American Chemical Society. | ||
Beyond differences in catalyst structure, the electrolyte composition also plays a substantial role in dictating the reaction mechanism. Specifically, electrostatic interactions between reactants/intermediates with ions in the double layer, hydrogen bonding, and local pH all alter the free energy landscape of electrocatalytic reactions. To investigate these effects within glycerol oxidation on Pd catalysts, operando IR spectroscopy was combined with electrochemical analysis.48 Here, the surface reaction mechanism of Pd nanoparticles anchored on carbon nanotubes in 0.1 M NaOH or 0.1 M KOH was studied. In KOH, the overall spectral changes, especially for C1 species such as carbonate, were less pronounced than in NaOH, indicating a lesser propensity to fully oxidize glycerol in KOH. The higher activity in NaOH was speculated to derive from a complexation effect in which Na+ ions interact with the organic reactants and intermediates to stabilize them on the Pd surface and thus promote their complete oxidation.
Sum frequency generation spectroscopy
Sum frequency generation (SFG) spectroscopy utilizes pulses of visible and infrared radiation to generate an SFG signal with the combined frequency of the two incident light pulses. Due to the selection rules of SFG, typically only signals at interfaces give rise to a substantial SFG signal, rendering this technique surface-sensitive.49 This would ideally be leveraged to detect transient surface intermediates that do not build up in the bulk solution and therefore provide unique insights into an electrocatalytic process.SFG was recently used to investigate HMF oxidation on Ni3N electrocatalyst surfaces in 1.0 M KOH.50 As a function of systematically increasing potential or as a function of time at a fixed potential, spectral signatures of HMF gave way to those of HMFCA and finally the spectrum resembled that of FDCA (Fig. 8a and b). This work elucidated that HMF oxidation on NiN3 proceeds via first the aldehyde oxidation, followed by the alcohol oxidation on HMF.
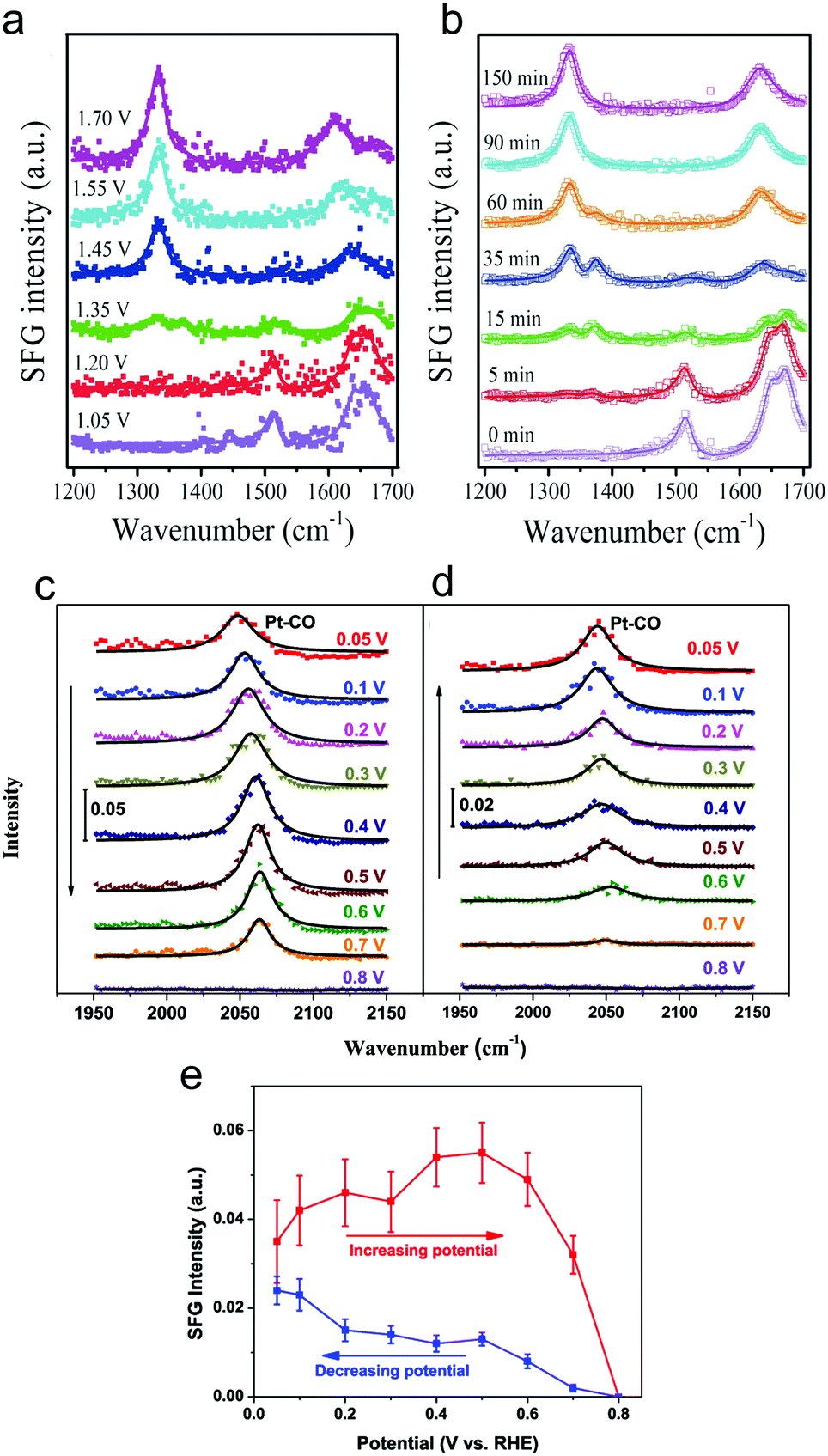 | ||
| Fig. 8 Operando SFG spectra of HMF oxidation on Ni3N catalyst surfaces after 90 minutes at select potentials (referenced vs. RHE) (a) or as a function of time at 1.45 V (b). CO was detected on polycrystalline Pt surfaces at low potentials and was more prevalent in the forward scan (c) than in the reverse scan (d). The intensities of the CO band as a function of potential are plotted in (e). Reprinted with permission from ref. 44 (N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903) and ref. 45 (Y. Liu, W. Yu, D. Raciti, D. H. Gracias and C. Wang, J. Phys. Chem. C, 2019, 123, 426–432). Copyright John Wiley and Sons 2020 and American Chemical Society 2019. | ||
The oxidation of glycerol in 0.1 M HClO4 electrolyte on polycrystalline Pt surfaces was investigated to extract insights into the surface catalytic process.51 CO was observed at weakly oxidizing potentials (0.05 V vs. RHE), indicating a dissociative adsorption and C–C bond cleavage occurred at low potentials (Fig. 8c–e). However, the low selectivity towards carbonate at low potentials was attributed to sluggish oxidation of the surface adsorbed CO and C2 fragments. In contrast, at high (more positive) potentials, partial oxidation of glycerol became more favourable as C–C bond cleavage was minimized and thus, less surface-bound CO was present.
Looking ahead
Through extensive efforts in understanding surface reaction mechanisms in CO2 reduction, the community is beginning to develop catalytic systems that are selective towards both C1 products as well as more complex C2 species. One can envision that a similar maturation process can be attained in the domain of electrochemical biomass valorization, with operando spectroscopy shedding light on how selective oxidation and reduction reactions can be carried out. Here, molecular systems can also play a role in serving as catalysts or as agents to modulate the reaction free energy landscape through secondary coordination sphere effects. In addition to the biomass platforms discussed in this article, the selective electrochemical transformation of lignin to value-added chemicals is a substantial challenge with significant payoff that stands to benefit from the insights derived through operando spectroscopy.52Beyond applications in biomass valorization, operando spectroscopy stands to play a key role in accelerating the electrification of the chemical industry. Parallel to the examples above, many commodity and fine chemicals can be generated through raw feedstocks and renewable electricity as opposed through thermochemical routes. The intricate knowledge of surface catalytically active sites, adsorbed intermediates, and reaction landscapes are important to the development of such systems. To this end, a notable work utilized Raman spectroscopy to probe the surface structure of Pd electrocatalysts in the oxidation of ethylene to ethylene glycol.53 Further, operando IR spectroscopy helped elucidate how the different surface facets of Pt feature varying favoured reaction pathways of acetone reduction and in general, how the coordination number of Pt surface sites dictates the free energy landscape for the reduction of aliphatic ketones.54 Coupling reactions, such as the electrosynthesis of dimethyl carbonate55 and diphenyl carbonate56 from CO, methanol and phenol building blocks have been investigated with operando IR spectroscopy. In the latter example, IR experiments illustrated that diphenyl carbonate could form either from methanol, CO and phenol, or also from dimethylcarbonate and phenol on gold surfaces (Fig. 9a and b). Thus, these results revealed that diphenyl carbonate was formed via a dimethyl carbonate intermediate via the oxidative coupling of methanol and CO, and the methoxy groups of the dimethyl carbonate were then replaced with the phenoxy groups from phenol (Fig. 9c). While these works represent a promising start, there is much more to investigate.
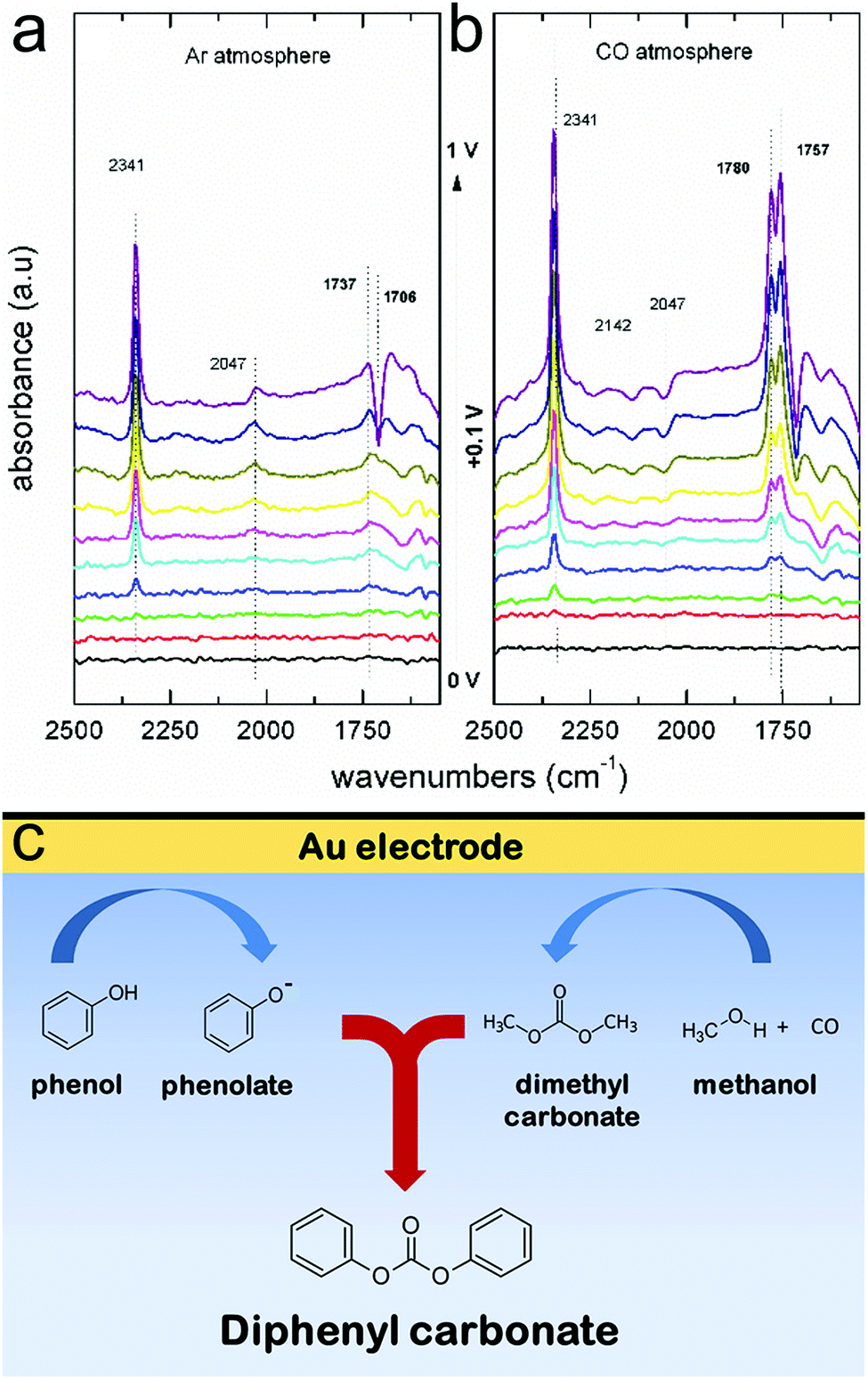 | ||
| Fig. 9 IR spectra of Au electrode in a methanol and phenol solvent, in the absence (a) and presence (b) of CO. The spectroscopic data are interpreted to suggest that diphenyl carbonate forms first through a surface-generated dimethyl carbonate intermediate, which itself forms through oxidative coupling between methanol and CO (c). Reprinted with permission from ref. 50 (M. C. Figueiredo, V. Trieu, S. Eiden, J. Heijl and M. T. M. Koper, ACS Catal., 2018, 8, 3087–3090). Copyright 2018 American Chemical Society. | ||
It is worth noting that while this feature article focuses on vibrational spectroscopy, there are several other powerful techniques that can provide complementary insights to fill in key knowledge gaps. Transmission electron microscopy (TEM) instrumentation has advanced to the point where TEM can be applied to directly probe electrocatalyst dynamics under an applied bias.57Operando TEM, conducted with liquid cells encapsulated with graphene or silicon nitride windows, has been instrumental in enabling the community in understanding phenomena ranging from battery behaviour58 to electrocatalyst reconstruction.59 A similar line of insights can be extracted from biomass valorization electrocatalysts.
X-ray based techniques, namely X-ray absorption near edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and X-ray photoelectron spectroscopy have been key in the electrocatalysis community. By providing information on catalyst electronic structure and coordination environment with various degrees of surface/bulk sensitivity, their use has permitted the formation of concrete structure–activity relationships.60–64 There is much room in the field of biomass valorization to form the same links.
Conclusions
In all, as the world looks towards expanding the use of renewable energy sources, the implementation of electrochemical technologies to valorize biomass to fuels and value-added chemicals is gaining traction. As this area is far less mature than established technologies operating at an industrial scale, many fundamental insights have yet to be made to advance biomass valorization to economic viability.To this end, this feature article highlights the utility of operando spectroscopic techniques that provide mechanistic information regarding catalyst dynamics, reaction intermediate identity, electrolyte effects and more. The derived knowledge aims to accelerate the design of next generation catalytic systems while also being translatable to other reaction schemes such as organic electrosynthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. K. and N. H. acknowledge the NSERC Grant R-N401-02839.Notes and references
- J. G. Olivier, J. A. Peters and G. Janssens-Maenhout, Trends in global CO2emissions 2012 report, Netherlands, 2012.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- A. D. Handoko, S. N. Steinmann and Z. W. Seh, Nanoscale Horiz., 2019, 4, 809–827 RSC.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 21, 749–770 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- T. J. Farmer and M. Mascal, Introduction to Chemicals from Biomass, John Wiley & Sons, Ltd, 2014, pp. 89–155, DOI:10.1002/9781118714478.ch4.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: volume I – results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab., Golden, CO (US), United States, 2004 Search PubMed.
- J. Carneiro and E. Nikolla, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 85–104 CrossRef CAS PubMed.
- S. S. Yazdani and R. Gonzalez, Curr. Opin. Biotechnol., 2007, 18, 213–219 CrossRef CAS PubMed.
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef PubMed.
- B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- R.-J. van Putten, A. S. Dias and E. de Jong, in Catalytic Process Development for Renewable Materials, ed. P. Imhof, 2013 Search PubMed.
- K. Dalvand, J. Rubin, S. Gunukula, M. Clayton Wheeler and G. Hunt, Biomass Bioenergy, 2018, 115, 56–63 CrossRef.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- T. Issariyakul and A. K. Dalai, Renewable Sustainable Energy Rev., 2014, 31, 446–471 CrossRef CAS.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- N. P. Martínez, M. Isaacs and K. K. Nanda, New J. Chem., 2020, 44, 5617–5637 RSC.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- M. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202002680.
- K. Li and Y. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed.
- J. Li, Y. Zhang and N. Kornienko, New J. Chem., 2020, 44, 4246–4252 RSC.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS PubMed.
- N. Heidary, K. H. Ly and N. Kornienko, Nano Lett., 2019, 19, 4817–4826 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS.
- Y. Deng and B. S. Yeo, ACS Catal., 2017, 7, 7873–7889 CrossRef CAS.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- J. R. Ferraro, K. Nakamoto and C. W. Brown, Introductory Raman spectroscopy, Elsevier, United States, 2nd edn, 2003 Search PubMed.
- Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- N. Heidary and N. Kornienko, Chem. Commun., 2019, 55, 11996–11999 RSC.
- N. Heidary and N. Kornienko, Chem. Sci., 2020, 11, 1798–1806 RSC.
- C. Liu, M. Hirohara, T. Maekawa, R. Chang, T. Hayashi and C.-Y. Chiang, Appl. Catal., B, 2020, 265, 118543 CrossRef.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- H. Miyake, S. Ye and M. Osawa, Electrochem. Commun., 2002, 4, 973–977 CrossRef CAS.
- H.-F. Wang, Y.-G. Yan, S.-J. Huo, W.-B. Cai, Q.-J. Xu and M. Osawa, Electrochim. Acta, 2007, 52, 5950–5957 CrossRef CAS.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937–3945 CrossRef CAS PubMed.
- A. C. Garcia, Y. Y. Birdja, G. Tremiliosi-Filho and M. T. M. Koper, J. Catal., 2017, 346, 117–124 CrossRef CAS.
- A. C. Garcia, M. J. Kolb, C. van Nierop y Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, ACS Catal., 2016, 6, 4491–4500 CrossRef CAS.
- G. A. B. Mello, C. Busó-Rogero, E. Herrero and J. M. Feliu, J. Chem. Phys., 2018, 150, 041703 CrossRef PubMed.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- D. Hiltrop, S. Cychy, K. Elumeeva, W. Schuhmann and M. Muhler, Beilstein J. Org. Chem., 2018, 14, 1428–1435 CrossRef PubMed.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- Y. Liu, W. Yu, D. Raciti, D. H. Gracias and C. Wang, J. Phys. Chem. C, 2019, 123, 426–432 CrossRef CAS.
- M. Zirbes and S. R. Waldvogel, Curr. Opin. Green Sustain. Chem., 2018, 14, 19–25 CrossRef.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D.-H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C.-T. Dinh, J. Li, T.-T. Zhuang, F. Li, T.-K. Sham, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 14–22 CrossRef CAS.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243–250 CrossRef CAS.
- M. C. Figueiredo, V. Trieu, S. Eiden and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 14693–14698 CrossRef CAS PubMed.
- M. C. Figueiredo, V. Trieu, S. Eiden, J. Heijl and M. T. M. Koper, ACS Catal., 2018, 8, 3087–3090 CrossRef CAS PubMed.
- N. Hodnik, G. Dehm and K. J. J. Mayrhofer, Acc. Chem. Res., 2016, 49, 2015–2022 CrossRef CAS PubMed.
- Y. Yuan, K. Amine, J. Lu and R. Shahbazian-Yassar, Nat. Commun., 2017, 8, 15806 CrossRef CAS.
- Y. Li, D. Kim, S. Louisia, C. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194 CrossRef CAS PubMed.
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- N. Kornienko, J. Resasco, N. Becknell, C.-M. Jiang, Y.-S. Liu, K. Nie, X. Sun, J. Guo, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2015, 137, 7448–7455 CrossRef CAS PubMed.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 CrossRef.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |